
第一作者:张红(吉林大学材料学院研究员)
通讯作者:杨春成&鹿可&宋斌
通讯单位:吉林大学/安徽大学/苏州大学

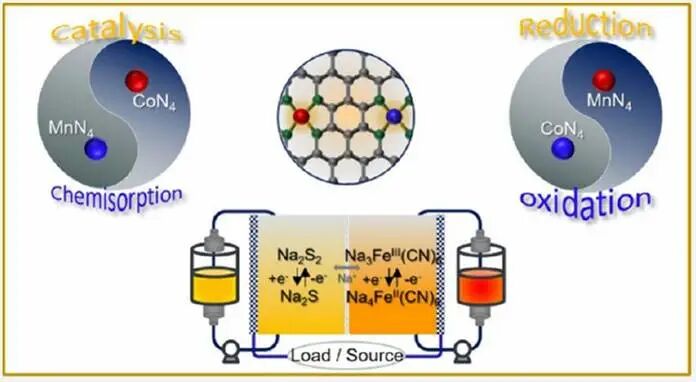
水系多硫化物/铁氰化物液流电池受限于多硫化物的缓慢氧化还原动力学和不可控的穿梭效应,导致活性物质利用率低,阻碍其实际大规模化应用。在此,吉林大学杨春成&张红团队开发了一种瞄定双单原子Mn和Co活性催化位点的氮掺杂功能性碳毡。研究揭示“优化的双单原子d带模型”可以调节Mn和Co的d带中心匹配Na2S2的LUMO和Na2S的HOMO之间的能隙,从而实现双向加速及协同催化Na2S2还原及Na2S氧化过程。此催化效果在正极电极液中铁氰化物的氧化还原过程也得到验证。组装的Fe-S液流电池在20 mA cm⁻²下表现出较高能量效率(76.4%)和优异的功率密度(119.3 mW cm⁻²),以及较好的循环稳定性(超过1000圈)及极低容量衰减率(每圈衰减0.0146%)。
TOC Figure
相关成果以“Mn−Co Dual-Metal Single-Atom Catalytic Sites for Boosted Redox Kinetics in Aqueous Polysulfide/Ferricyanide Flow Batteries”为题发表在Nano Letters期刊上。
感谢吉林大学张红研究员供稿!
本文所用
一体化液流单电池测试系统(YTH-1/LSB-1)
由武汉之升新能源有限公司提供

水系多硫化物液流电池(FBs)具有高安全性、低成本和环境友好的优点,是电网级储能的理想候选者。针对实际应用,研究人员探索了多种水系多硫化物氧化还原电对,如多硫化物–溴、多硫化物–碘化物、多硫化物–锰酸盐、多硫化物–空气以及多硫化物–亚铁氰化物等体系。其中,S22-/S2-和Fe2+/Fe3+电对因其在水中的高溶解度和成本效益,成为构建高能量水系Fe-S FBs的优选活性物质。如图1a所示,Na2S2溶液作为负极电解液,Na3Fe(CN)6溶液作为正极电解液。在温和的水系电解液中配对S22-/S2-(-0.55 V)和Fe3+/Fe2+(0.35 V)氧化还原电对,Fe-S全电池可实现约0.9 V的理论平均工作电压。此外,具有S22-/S2-双电子反应的Fe-S FBs展现出高达53.6 Ah L-1mol-1的理论体积容量。
尽管Fe-S FBs被广泛探索,其实际应用仍受限于活性物质的有限再利用和低效的能量输出。多硫化物的严重穿梭效应导致Fe-S FBs循环性能差,难以与钒基和锌基FBs相媲美。此外,S22-/S2-电对在裸碳界面上固有的缓慢动力学会导致严重的电化学极化、不受控的歧化反应、库仑效率下降和电化学可逆性差(如图1b)。因此,需要加强基质与活性物质之间的相互作用,以加速多硫化物的转化并抑制其穿梭,实现长期稳定运行。近年来,电极改性的催化工程被提出以调控多硫化物转化的氧化还原动力学,可提供活性位点增强吸附和催化能力,并有效缓解穿梭效应(图1c)。然而,相关电池的循环性能在某些方面仍远未达到实际应用要求。单原子催化剂(SACs)因其在多硫化物化学吸附中理论100%的原子利用效率而受到广泛研究。此外,S物种的p带中心与SACs的d带中心之间的小能带隙有利于快速界面电子动力学和高效的硫转化催化。因此,开发基于单原子的催化基质是提高S22-/S2-氧化还原反应活性、降低电化学极化、提高能量效率和延长Fe-S FBs使用寿命的可行途径。
在此,该团队设计了一种核壳结构的催化石墨碳毡,MnN4和CoN4位点组成的多孔碳化封装组成(表示为MnCoNC GF)。DFT模拟结果显示,MnN4和CoN4中心对多硫化物和Na2S物种表现出更强的化学吸附作用(见图1d),并且CoN4位点可以显著降低Na2S的氧化能垒(图1e)。Mn和Co的d带心分别上下移动,以优化Na2S2和Na2S的LUMO和HOMO之间的相关能隙,从而产生强大的协同效应,使MnCoNC基底同时双向加速Na2S2还原和Na2S氧化。此外,MnN4部分可以显着加速Na3Fe(CN)6和Na2S2还原过程,而CoN4位点促进了Na4Fe(CN)6和Na2S再氧化反应(图1f和1g)。Mn-Co协同催化位点表现出高电催化性能,可增强硫物质的再利用并调节Na3Fe(CN)6/Na4Fe(CN)6氧化还原对的可逆转化。因此,基于MnCoNC-GF的水性Fe-S FB表现出优异的电化学性能和长循环稳定性。
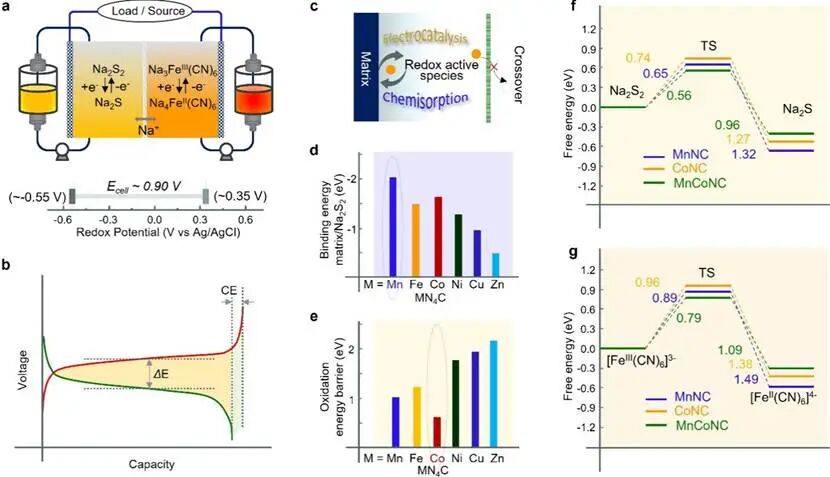
图1 Fe-S液流电池系统的电化学性能和密度泛函理论计算。(a)水系多硫化物液流电池中的反应以及Na2S2/Na2S和Na3Fe(CN)6/Na4Fe(CN)6氧化还原对的理论氧化还原电位。(b)Fe-S液流电池放电−充电电压曲线的示意图,突出显示液流电池系统中仍需解决的关键问题。(c)物理化学限制效应与电催化效应协同调控硫氧化还原过程物种的示意图。(d,e)Na2S2的吸附能及Na2S在不同基底(包括MnN4C、FeN4C、CoN4C、NiN4C、CuN4C和ZnN4C)上的氧化能垒。预测还原反应的自由能图,(f) Na2S2/Na2S和(g) Na3Fe(CN)6/Na4Fe(CN)6在不同基底上的反应。
1. MnCoNC-GF催化剂的设计与合成
设计了由多孔碳封装双原子MnN4和CoN4位点组成的核壳结构催化石墨碳毡(MnCoNC-GF)。如图2a所示,制备步骤主要包括:以过硫酸铵为引发剂,在原始碳毡表面原位共聚苯胺和吡咯;Mn2+/Co2+阳离子被互连聚合物框架中的杂原子(如N)捕获,形成PANi-PPy-Co2+/Mn2+-GF;随后对聚合物前体进行热处理碳化,得到高度石墨化的多孔碳层;最后通过酸蚀刻和二次热处理获得目标功能化碳毡。MnNC-GF和其他对照基质也使用类似程序制备。优化后的碳基质通过调整前驱体确定,吡咯和苯胺的共添加可增强杂原子和金属原子掺杂含量。连续的两步酸浸过程对于去除松散附着的Mn/Co组分、修复碳微结构并同时提高原子掺杂水平(Mn: 2.47 wt %, Co: 2.04 wt %)至关重要。图2b和2c显示了MnCoNC-GF的核壳形貌,具有约5 nm厚的多孔碳层。高分辨TEM图像(图2c, 2d)显示出清晰石墨晶格条纹和丰富的元素原子点。元素映射(图2e)证明了Co、Mn和N物种在整个碳框架中的均匀掺杂。更重要的是,通过原子分辨率HAADF-STEM和EELS鉴定出碳层内嵌入的丰富原子级金属位点。EELS点谱(图2f)证实了埃米尺度内N与Mn或Co的共存,表明孤立的单原子位点由碳框架内的N原子锚定。Mn和Co K边XANES和EXAFS表征进一步揭示了单核中心的局域结构和配位信息。图2g中的实验XANES谱证实MnCo-NC-GF中Mn K边的预边位于MnPc和Mn箔之间且更接近MnPc,表明MnCo-NC中Mn的氧化态接近+2,其配位环境类似于明确的MnN4化学结构。此外,MnCoNC-GF中的Mn K边在约1.45 Å处显示主峰(图2h),对应于Mn-N键。未观察到明显的Mn-Mn、Mn-Co和/或Mn-O配位峰,根据拟合结果(CNMn-N = 3.9 ± 0.3),MnCo-NC中的主导Mn-N结构很可能是MnN4。Co K边的XANES谱也显示出类似结果,位于约1.40 Å的主特征峰对应于Co-N配位(图2i)。EXAFS拟合进一步表明Co表现出平均4.1的Co-N配位数,证明成功构建了具有MnN4/CoN4配位结构的MnCoNC(图2j)。小波变换EXAFS进一步验证了这些观察结果(图2k, 2l)。
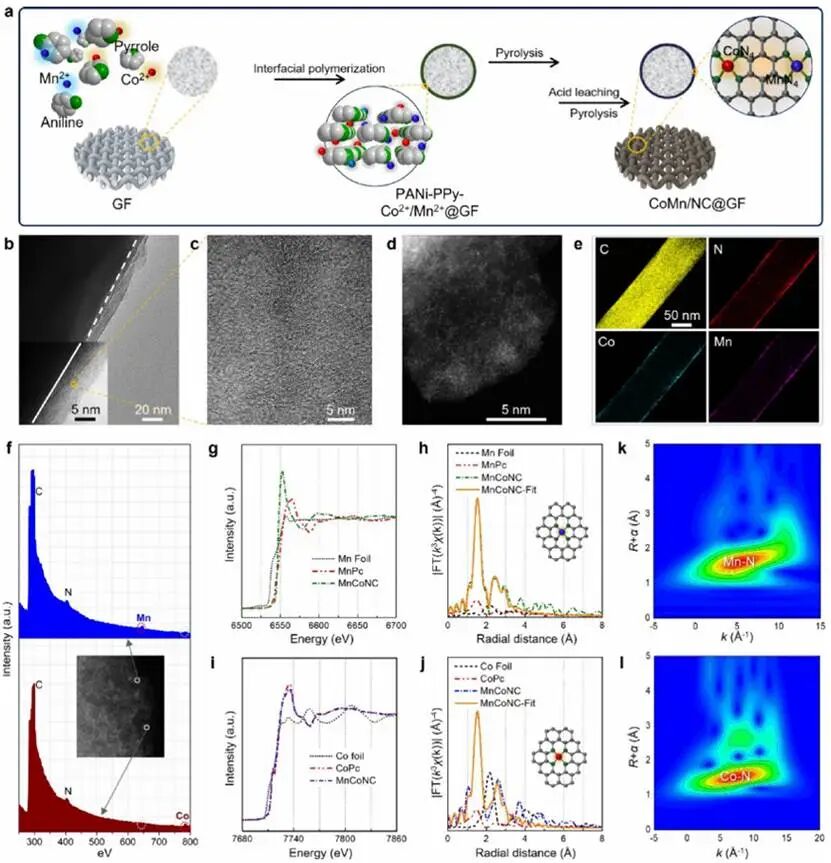
图2 MnCoNC-GF的设计示意图。(a) MnCoNC-GF电极合成的主要步骤。(b,c) 透射电子显微镜(TEM)图像,显示MnCoNC-GF的壳层结构形态及无定形MnCoNC壳层。(d) 原子分辨率HAADF-STEM图像,(e) 元素映射图像,以及(f) MnCoNC-GF的电子能量损失谱(EELS)分析。(g) Mn K-edge和(i) Co K-edge XANES光谱。(h,j) Mn/Co的傅里叶变换(FT)-EXAFS光谱,以及傅里叶变换实验EXAFS光谱与基于MnN4/ConN4模型结构模拟的拟合光谱的比较。(k,l) MnCoNC-GF的EXAFS信号的小波变换。
2. 多硫化物锚定与协同催化机制
通过Na2S2捕获测试评估了主体材料与多硫化物的相互作用。选择不同的碳毡研究其对多硫化物物种的吸附能力。如图3a所示,含有MnCoNC-GF的Na2S2水溶液比对照电解液表现出最明显的褪色,几乎变为无色。Na2S2溶液的可见褪色过程证明了MnCoNC-GF与Na2S2之间的强相互作用。定量UV-Vis结果(图3b)显示,与裸Na2S2溶液相比,添加GF和处理后的GF后吸收峰强度降低,突出了基质依赖的多硫化物锚定能力。S22-的吸收峰强度按GF, NC-GF, CoNC-GF, MnNC-GF, MnCoNC-GF的顺序递减。值得注意的是,Mn催化中心的吸附能力强于Co位点,这与理论计算结果一致。Mn/基质与Na2S2之间的结合能高于Co/基质,进一步表明Mn热点对多硫化物稳定性的主要增强有贡献。穿梭电流测量用于量化受抑制的穿梭效应。MnCoNC-GF组装的电池具有最小的穿梭电流密度,约2.2 μA cm-2,表明双原子Mn-Co催化位点的协同效应有利于增强吸附能力和抑制Fe-S FBs内的多硫化物穿梭。
为深入研究Mn/Co原子与多硫化物的相互作用,对MnCoNC-GF电极在多硫化物–基质相互作用前后进行了XPS分析。相应地,吸附Na2S2溶液后,MnCoNC-GF的Mn 2p(图3c)和Co 2p(图3d)峰向较低结合能移动,其中Mn 2p峰(0.46 eV)比Co 2p峰(0.1 eV)显示出更大的位移,证明Mn位点对Na2S2分子的亲和力显著高于Co中心。因此,电荷重新分布促进了后续的氧化还原动力学。此外,MnNC表现出较高的d带中心(εd),接近基底Na2S2的LUMO(图3e)。与LUMO的微小能隙使得电子从Mn 3d向Na2S2的LUMO转移更快,促进了S-S键的快速断裂以实现快速还原。CoNC的εd明显接近Na2S的HOMO,因此Na2S可以容易地将电子转移到CoNC。如图3f示意图所示,当Mn和Co耦合形成MnCoNC时,Mn和Co的εd分别上移和下移,以优化Na2S2的LUMO和Na2S的HOMO之间的相关能隙,然后协同催化效应使MnCoNC基质能够同时双向加速硫氧化还原。
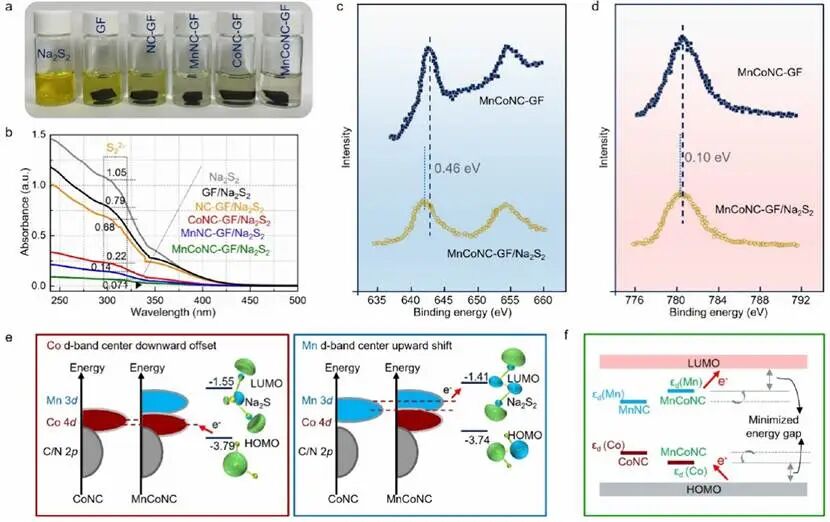
图3 功能性基质对Na2S2物种的吸附及电催化效应。(a) 不同样品可视化吸附试验的光学图像。(b)在(a)吸附试验前后Na2S2溶液的对应紫外–可见吸收光谱。高分辨率XPS(c) Mn 2p和(d) Co 2p区域光谱在MnCoNC-GF材料与钠多硫化物相互作用前后的比较。(e)单金属单原子系统与双金属单原子系统中d带中心位置的比较,以及催化剂d带中心能级示意图和相关硫化物种的LUMO/HOMO能级。(f) “优化d带模型”的示意图。
3. 电化学性能与电池验证
为了探索基质依赖的氧化还原活性的电催化和动力学行为,首先在三电极配置中进行循环伏安(CV)测试(图4a, 4b)。相比之下,MnCoNC-GF显示出最高的电流密度和最低的峰电位分离,表明其具有优异的氧化还原动力学和高可逆性。值得注意的是,与CoNC-GF电极相比,MnNC-GF的还原峰发生正移,表明MnN4是加速Na2S2还原为Na2S的主要热点。相反,CoN4中心促进高效的Na2S再氧化,这由负移的氧化峰所证明,与DFT结果一致。此外,在不同扫描速率下对S22-/S2-进行CV以研究不同电极的传质行为和反应动力学,峰值电流与扫描速率符合Randles-Sevcik方程。计算得出MnCoNC-GF电极中阳离子的扩散系数为7.23 × 10-6cm2 s-1,高于裸GF和其他处理的GF。
MnCoNC-GF在Na2S2/ Na2S氧化还原过程中具有最低的Tafel斜率(还原53 mV dec-1,氧化13 mV dec-1)(图4c),这进一步表明Co和Mn中心协同增强了化学吸附多硫化物物种的双向反应动力学。在阴极液腔室中也观察到类似的协同趋势。如图4b所示,MnCoNC-GF电极上Fe3+/Fe2+氧化还原峰之间的电位差最小。计算了Na3Fe(CN)6/Na4Fe(CN)6氧化还原过程的Tafel曲线,清晰可见MnCoNC-GF电极对还原峰和氧化峰均表现出最低的Tafel斜率(分别为66和150 mV dec-1)(图4d)。此外,获得的奈奎斯特曲线显示MnCoNC-GF电极具有最低的电荷转移电阻,这与CV分析的结论一致。所有这些结果表明Fe3+/Fe2+氧化还原峰的趋势及相关氧化还原动力学的双向加速与S22-/S2-电对相似(图4e)。基于不同浓度电解液的稳定性和电化学性能测试,选择了3.0 M的钒浓度(nVOCl3:V2O5=1.9:0.55)、2.8M的SO42-和5.7 M的Cl–进行单电池性能测试。使用裸GF和改性GF分别作为正极和负极基质组装Fe-S FBs进行充放电测试。如图4f所示,在20 mA cm⁻²下观察到明确的充放电电压平台。未处理GF基电池的过电位远高于改性GF基电池,与MnCoNC-GF组装的液流电池相比降低了46%以上(图4g)。组装MnCoNC-GF的电池显示出最高的容量(50.1 Ah L-1,理论值的93.2%),高于其他电极的库仑效率(CE),并大大降低了充放电过电位,验证了引入催化效应的积极影响。
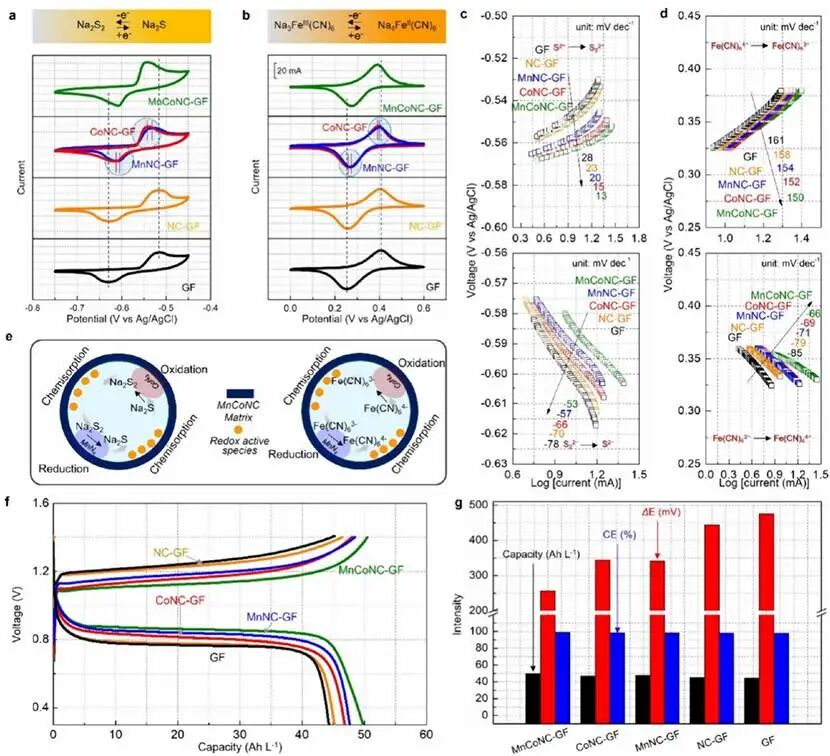
图4 电催化可调控的Na2S2/Na2S和Na3Fe(CN)6/Na4Fe(CN)6氧化还原行为。(a) 在扫描速率为5 mV s-1时,0.1 M Na2S2和(b) Na3Fe(CN)6在不同基质中的循环伏安曲线,突出了不同宿主对氧化还原过程的促进作用。对应于(c) Na2S2还原和Na2S2氧化,以及(d) Na3Fe(CN)6还原和Na4Fe(CN)6氧化的塔菲尔图。(e) 设计MnCoNC-GF电极双向催化性能的示意图。(f)不同电极材料Fe-S液流电池在20 mA cm-2电流密度下组装后充放电曲线的比较。
分别使用非原位XPS和原位UV-vis光谱进一步监测了阴极液和阳极液的化学演变。在充电过程中,制备的棕色Na2S2溶液发生电化学还原,导致溶液颜色变为浅黄色。这与图5a中的非原位XPS结果一致,其中162.4和163.6 eV的特征峰属于Na2S2,其强度在充电过程中逐渐降低。同时,Na2S的特征峰在162.3和163.5 eV处逐渐出现,并在电池充电结束时达到最大强度。随后的放电过程揭示了这些多硫化物峰变化的可逆趋势。在充电过程中,阳极的Na4[Fe(CN)6]逐渐氧化为Na3[Fe(CN)6]。结果,Fe2+在709.2和722.9 eV的峰逐渐减弱并移至710.7和724.2 eV,如图5a中电位依赖的非原位XPS谱所示。同时,随着电池充电深度的增加,Fe3+峰强度显著增加。在充电过程结束时,当Fe2+处于100%充电状态时,所有归属于Fe2+的峰消失,而与Fe3+相关的峰达到最大强度。原位UV-vis表征进一步证实了上述结论(图5b)。随着放电进行,阴极液中Fe(CN)64-的峰强度降低,同时Fe(CN)63-峰强度增加。然后在随后的再充电过程中,Fe(CN)63-的峰强度降低。对于阳极液,可以检测到S22-和S2-的特征峰,其峰强度变化趋势与阴极液非常相似,表明Na2S2↔Na2S转化的可逆性。所有这些演变在放电过程后都能恢复到原始状态,证明了Na3Fe(CN)6阴极液的结构稳定性和电化学可逆性。
对MnCoNC-GF组装的Fe-S FBs进行了倍率性能测试,电流密度从20 mA cm-2到100 mA cm-2。观察到,即使测试电流增加了5倍,电池仍保持了超过76%的高容量保持率(图5c)。如图5d所示,极化曲线和功率密度曲线清楚地表明MnCoNC-GF电极具有最高的功率密度119.3 mW cm-2,优于GF (48.0 mW cm-2)、NC-GF (51.5 mW cm-2)、Co-NC-GF (67.7 mW cm-2)和Mn-NC-GF (93.7 mW cm-2)。此外,图5e概述并比较了Fe-S FBs在所有电流密度下的CE、VE和EE趋势。MnCo-NC-GF在所有电流密度下均表现出高于其他对照电极的EE、VE和CE值,同时充放电过电位大大降低。MnCo-NC-GF的CE在所有电流密度下均保持在约99.5%以上,而EE和VE分别从20 mA cm-2时的76.4%下降到100 mA cm⁻²时的58.3%,以及从20 mA cm-2时的76.8%下降到100 mA cm-2时的58.4%。观察到的VE和EE下降趋势通常归因于较高电流密度下电池过电位的增加。研究了MnCoNC-GF组装的Fe-S液流电池系统的长期循环性能,表现出优异的循环稳定性,平均CE超过99%,1000次循环后容量保持率高达85.4%(图5f)。研究表明多硫化物穿梭相当缓慢,计算的质量损失率仅约4.0%。催化基质的优化可以有效抑制Fe-S FBs中的多硫化物穿梭。循环后,微观结构的原子级分散位点和电化学响应几乎没有变化。此外,XPS分析也证实了原子级Mn和Co位点的稳定性以及无聚集的Mn/Co物种。因此,双原子催化位点的设计可能有利于同时提高电化学性能和结构稳定性。与报道的其他多硫化物FBs系统和其他最近提出的RFB系统相比,所提出的Fe-S FBs表现出显著更低的材料成本和优异的能量/功率密度以及稳定的循环性能。
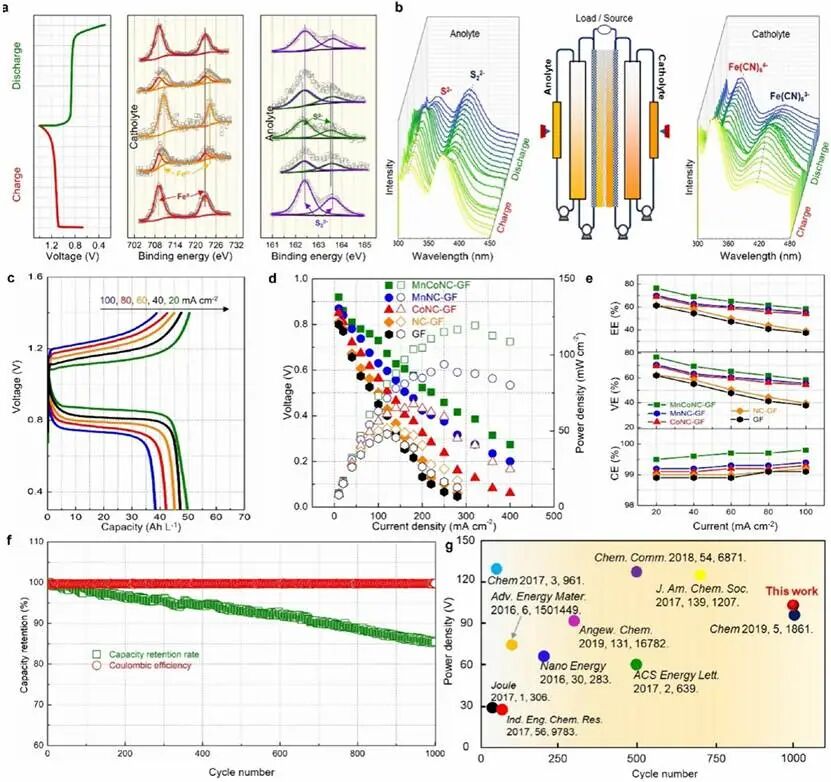
图5 原位和半原位表征。(a) 液流电池在充放电过程中阴极液和阳极液的半原位XPS光谱。(b) 阴极液和阳极液在放电和充电过程中原位UV-vis表征系统的示意图,以及相应的UV-vis光谱。(c) 不同电流密度下组装的MnCoNC-GF Fe-S液流电池的电压曲线。(d) Fe-S液流电池的极化曲线。(e) 不同基质组装的Fe-S液流电池在不同测试电流密度下的CE、VE和EE比较。(f) 在60 mA cm-2电流密度下,MnCoNC-GF 组装的Fe-S液流电池系统的循环性能。(g) Fe-S液流电池与其他代表性RFB的功率密度比较。
本文制备了具有MnN4和CoN4双催化活性位点改性的碳石墨毡作为电催化反应器用来双向催化水系Fe-S FBs中的氧化还原动力学,并有效缓解多硫化物穿梭效应。逐步建立的“优化的d-band模型”证明了Mn和Co的d带中心分别上移和下移,以优化Na2S2的LUMO和Na2S的HOMO之间的能隙,协同催化效应使MnCoNC-GF功能性碳毡能够在电池动态放电和充电循环过程中双向加速Na2S2还原和Na2S氧化。相应地,展示了一种低成本、长循环的Fe-S液流电池,其在20 mA cm-2下具有76.4%的高能量效率,119.3 mW cm-2的优异功率密度,以及1000次循环中每循环仅0.0146%的极低容量衰减率。这项工作为设计高效、稳定的双功能催化剂以推动低成本液流电池的实际应用提供了重要参考。
Mn−Co Dual-Metal Single-Atom Catalytic Sites for Boosted Redox Kinetics in Aqueous Polysulfide/Ferricyanide Flow Batteries, 2025, Nano Letters, https://doi.org/10.1021/acs.nanolett.5c02013
第一作者:张红,吉林大学材料科学与工程学院,研究员,2021年在上海交通大学获得博士学位;期间在美国Northern Illinois University/Argonne National Laboratory联合培养,2021-2024年先后在中国科学技术大学和哈尔滨工业大学从事博士后及助理教授职位;2024年12月入职吉林大学材料科学与工程学院。研究聚焦于:能源存储与过程强化,水系锌电,金属硫电池等,在Angew. Chem.、CCS Chem.、Energy Environ. Sci.、Nano Lett.等期刊上发表SCI论文50余篇。
通讯作者:鹿可,安徽大学教授,江苏省姑苏创业领军人才。2018年在山东大学获得博士学位(导师:马厚义教授);2017年-2018年,在同济大学进行固态锂/钠电池项目博士联培(导师:黄云辉教授/罗巍教授);2018年-2020年,在美国Northern Illinois University/Argonne National Laboratory从事博士后研究;2020年9月入职安徽大学。研究工作聚焦于(固态)金属硫电池体系的关键材料与界面基础问题,致力于揭示硫转化的催化调控,构建高能量实用型金属硫电池器件。发表SCI论文105篇,H-index为38。课题组主页:https://www.x-mol.com/groups/look
通讯作者:杨春成,吉林大学材料学院教授/副院长,吉林省首批“长白山学者”特聘教授,吉林大学“唐敖庆学者”领军教授。研究聚焦于:高性能钠/钾离子电池和燃料电池用金属基复合电极材料,发表SCI论文100余篇,包括Mater. Sci. Eng. R., Nat. Commun., Matter, Nano Lett.等,授权国家发明专利21件。任中国金属学会材料科学分会第八届委员会委员。
延伸阅读(张红团队近期论文合集):