
第一作者:韩明君
通讯作者:张晨阳&李洁
通讯单位:中南大学
成果简介
多酸(POMs)在下一代高能量密度氧化还原液流电池(RFBs)中展现出卓越的多电子转移能力,但其可操作的充电状态(SoC)通常受限于质子饥饿条件下普遍存在的高还原亚稳态而低于33.3%水平。该研究通过为[P2W18O62]6⁻({P2W18})团簇建立质子耦合电子转移(PCET)模型,揭示了氧位点的质子化可通过协同质子–电子转移(CPET)稳定还原的钨位点。Marcus理论与DFT计算相结合,量化了区域选择性CPET过程的热力学驱动力与动力学能垒,而通过原位pH监测与原位拉曼光谱进一步证实了这一质子耦合的可逆氧化还原机制。基于这些发现,该研究设计了高质子活性H6{P2W18}负极电解液,与VOSO4基正极电解液配对组成HPVB液流电池,并采用直接和逐步充放电策略,从而实现了H6{P2W18}负极电解液稳定的全SoC充放电循环。HPVB电池取得了前所未有的性能:在0.3M H6{P2W18}负极电解液66.7% SoC下循环600次(超过1020小时)无明显容量衰减,期间保持95.04 Ah L⁻1的放电容量;在0.3M H6{P2W18}负极电解液100% SoC下放电容量达141.75 Ah L⁻1;在0.5M H6{P2W18}负极电解液100% SoC下,放电容量创纪录地达到236.03 Ah L⁻1,能量密度达239.02 Wh L⁻1。该研究通过将CPET机理见解转化为可操作的电解质设计,释放了{P2W18}的稳定可逆全SoC,为通往高能量密度POM基氧化还原液流电池建立了一条普适性路径。
相关成果以“Unlocking Full State-of-Charge of Polyoxometalate for High-Energy-Density Redox Flow Batteries via Concerted Proton-Electron Transfer”为题发表在Angewandte Chemie International Edition期刊上。
感谢中南大学李洁&张晨阳教授(第一作者:韩明君)供稿!
本文所用
快拆型液流单电池测试系统(KCB-CZ)
由武汉之升新能源有限公司提供


研究背景
全球向可再生能源的加速转型预计将在2050年前供应近90%的电力,这已成为应对气候变化同时满足日益增长的能源需求的基石。然而,太阳能/风能发电的间歇性和波动性特性要求储能技术实现突破性创新,以确保电网可靠性并最大化可再生能源的利用率。在各种储能技术中,氧化还原液流电池(RFBs)因其本征安全性、通过解耦功率/容量配置实现的可扩展性、长循环寿命以及毫秒级响应特性,已成为大规模应用中最有前景的解决方案之一。
虽然商业部署的钒(VRFBs)、铁铬(Fe-Cr RFBs)和锌溴(Zn-Br RFBs)氧化还原液流电池日趋成熟,但科学界正积极开创下一代氧化还原活性电解质的分子水平工程。传统的单/双电子(n = 1~2)氧化还原对主导了超过95%的现有体系,主要包括过渡金属、卤素、多硫化物和有机化合物。然而,单/双电子转移反应所导致的低能量密度(15~50 Wh/L)限制了其进一步发展的潜力。因此,探索稳定且可逆的多电子(n ≥ 3)转移氧化还原活性对已成为RFBs创新的关键研究前沿。
多酸(POMs),特别是Keggin型([XM12O40]ⁿ⁻)和Dawson型([X2M18O62]ⁿ⁻)团簇,已成为多电子氧化还原化学中的领跑者,利用其独特的团簇级电子离域特性,通过Mⱽᴵ/Mⱽ氧化还原过程呈现超过12e⁻的转移能力。然而,研究表明,多酸的实际电解质利用率(此处用荷电状态SoC表示)仍被限制在低于33.3%的水平。低SoC的瓶颈普遍存在于各类多酸负极电解液中。2013年,Anderson等人首次将原子取代的[SiV3W9O40]7⁻({SiV3W9})用于对称RFB电解质,实现了{SiV3W9}负极电解液33.3%的SoC(相对于9e⁻理论容量的3e⁻转移)。随后,Liu等人于2016年开创性地使用H₆{CoW₁₂O₄₀}({CoW₁₂})团簇设计了对称氧化还原液流电池,实现了{CoW12}负极电解液33.3%SoC(4e⁻转移相对于12e⁻理论容量)。Chen等人于2018年首次在{P2W18}||Br RFB中采用Li6[P2W18O62](Li6{P2W18})作为负极电解液,建立了使用高还原态{P2W18}团簇进行能量存储的概念验证。然而,RFB的循环仅限于20个循环,在Li6{P2W18}负极电解液约88.9%的SoC下进行(放电容量和库仑效率相对于18e⁻理论容量换算),且其潜在的氧化还原化学机制仍未得到充分阐明。最近,Ai等人于2022年报道了在低温(-20 ℃)下使用H6[P2W18O62](H6{P2W18})团簇作为负极电解液(33.3%SoC,6e⁻转移相对于18e⁻理论容量)实现高功率密度氧化还原液流电池的进展,突出了其在宽温度范围内的稳健性和实用性。此外,[PW12O40]3⁻({PW12},SoC分别为33.3%和41.7%,4e⁻和5e⁻转移分别相对于12e⁻理论容量)、[PV14O42]9⁻({PV14},28.5%SoC,4e⁻转移相对于14e⁻理论容量)、以及[SiW12O40]4⁻和[BW12O40]5⁻({SiW12}和{BW12},16.7%SoC,2e⁻转移相对于12e⁻理论容量)也证实了这一点。因此,解锁多酸的全SoC以突破电解液低利用率这一普遍瓶颈,对于实现高能量密度RFBs将是一个关键的里程碑。
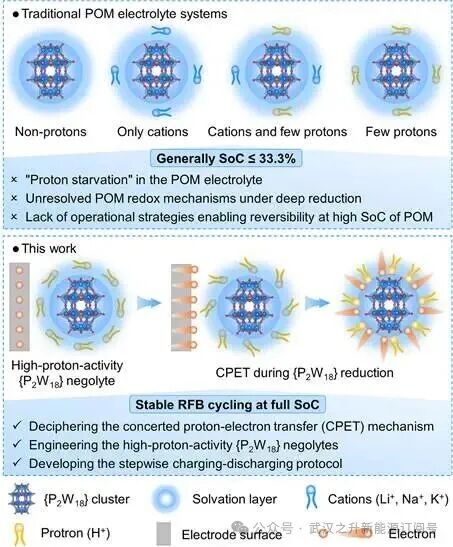
示意图1. 传统POM电解质体系与本工作之间的本质对比。
该研究指出,传统POM电解质体系的低利用效率主要源于深度还原条件下未阐明POM氧化还原机理,以及缺乏能够在高SoC下实现可逆性操作电解质策略。关键在于,POM还原过程中的“质子饥饿”条件限制了电荷存储与质子化的动态耦合,导致可达到的SoC通常低于33.3%(示意图1)。这些关键障碍从根本上限制了POM固有的多电子能力,导致实际电解质利用率远低于其理论潜力。为了克服这些限制,该研究建立了质子耦合电子转移(PCET)模型来解读{P2W18}氧化还原化学的结构–活性关系。该研究揭示了氧位点的质子化通过区域选择性协同质子–电子转移(CPET)过程稳定了还原的钨位点。这种深入的机理见解使得设计高质子活性的{P2W18}负极电解液并开发逐步充放电策略成为可能。由此得到的氧化还原液流电池进一步在H6{P2W18}负极电解液的高SoC(66.7%和100%)下实现了前所未有的稳定循环性能,为实现稳定可逆、耐用且高能量密度的POM-RFBs的全SoC提供了新路径。
核心内容
1.{P2W18}的电化学和结构特征
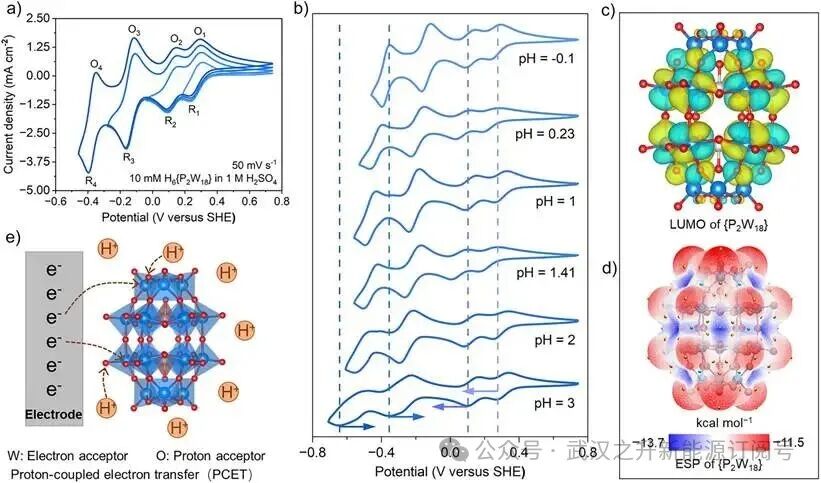
图1. {P2W18}的电化学和结构特征分析。(a)10 mM H₆{P₂W₁₈}在1 M H₂SO₄中于50 mV s⁻¹扫描速率下的分段循环伏安(CV)曲线。(b)10 mM H₆{P₂W₁₈}在不同pH介质中的CV曲线。(c){P₂W₁₈}的最低未占分子轨道(LUMO)分布。(d){P₂W₁₈}的静电势(ESP)图,表面的黄色和蓝色球体分别对应静电势的最大值和最小值点。(e){P₂W₁₈}团簇在电极界面的质子耦合电子转移(PCET)过程示意图。
对{P2W18}的循环伏安(CV)分析显示,通过分段电位窗口扫描可观察到四对高度可逆的氧化还原峰,如图1a所示,其中氧化峰(O1、O2、O3、O4)与还原峰(R1、R2、R3、R4)依次标注。O1/R1至O4/R4氧化还原特征峰电位分别为0.293 V/0.228 V、0.148 V/0.090 V、−0.115 V/−0.166 V和−0.350 V/−0.397 V(相对于标准氢电极,SHE)。这些氧化还原源于{P2W18}团簇中{W18O54}壳层内发生的累计约六个电子转移(顺序为1、1、2、2),具体对应于WVI/WV的氧化还原过程。各阶段的峰电位分离值(ΔEp)和峰电流比(Ipa/Ipc)证实了所有四个氧化还原阶段均具有优异的电化学可逆性。需要指出的是,{P2W18}的氧化还原电位在不同导电介质中表现出pH依赖的定向移动(图1b)。以pH=3(Na⁺主导体系)为参照:(1)在H⁺/Na⁺主导体系(pH=−0.1、1、2)中,R1和R2还原峰出现轻微的负移(ΔE=10~39 mV),而在H⁺缺乏/无Na⁺体系(pH=0.23、1.41)中则出现显著的负移(ΔE=69~102 mV),说明O1/R1和O2/R2氧化还原电对受H⁺浓度调控;(2)R3和R4还原峰随着pH降低(H⁺浓度升高)呈现显著的正移(ΔE=63~251 mV),表明O3/R3和O4/R4氧化还原电对具有较强的H⁺敏感性。综上,{P2W18}团簇的氧化还原过程显著受H⁺(质子)含量调控,推测这种H⁺依赖行为源于氧化还原过程中对抗衡离子及质子化的需求。通过CV和旋转圆盘电极(RDE)进一步研究了H6{P2W18}的氧化还原动力学(图S4、S5及表S2)。结果表明,其具有较高的扩散系数(D0=2.49×10⁻6cm2 s⁻1)和较快的电子转移速率常数(k0=8.9×10⁻4cm s⁻¹,高于V3⁺/V2⁺的3.9×10⁻5 cm s⁻1),说明其具备有利于电化学过程的氧化还原动力学。
{P2W18}团簇的活性位点可解耦为以W为中心的“电子活性位点”和以O为基础的“质子化活性位点”。前线分子轨道分析(图1c)表明,{P2W18}中最低未占分子轨道(LUMO)的空间分布主要定域于W中心,这归因于其丰富的W 5d空轨道所导致的缺电子特性。这些缺电子的W位点通过d轨道适应性电子填充实现可逆的电子存储/释放:WVI(5d0)+ e⁻ ⇌ WV(5d1)。此外,静电势(ESP)分析表明,{P2W18}团簇(电荷:−6)的亲核区域对其质子化反应至关重要。如图1d所示,计算得到的ESP等值面呈现出−13.7至−11.5 kcal/mol的定量色阶范围,并具有三个特征区域。ESP最小值(蓝色)集中于{P2W18}中相邻三金属氧簇(W3O15)的中间区域,对应最高电子密度;相对地,ESP最大值(红色)分布于端氧(W=Oₜ),而桥氧(W−Ob−W)区域(白色)介于两者之间,反映Oₜ处电子密度略低于Ob。Oₜ与Ob之间狭窄的ESP差异表明二者具有相当的质子亲和力,因此{P2W18}团簇的Ob和Oₜ位点均可有作为质子化反应的氢键受体的可能性。
因此,图1e进一步阐释了{P2W18}在电极界面还原时可能发生的质子耦合电子转移(PCET)过程,其中以W为中心的“电子活性位点”与以Oₜ/Ob为中心的“质子化活性位点”之间的协同作用,可能共同促成高效的多电子转移反应。
2. {P2W18}氧化还原过程的质子耦合电子转移
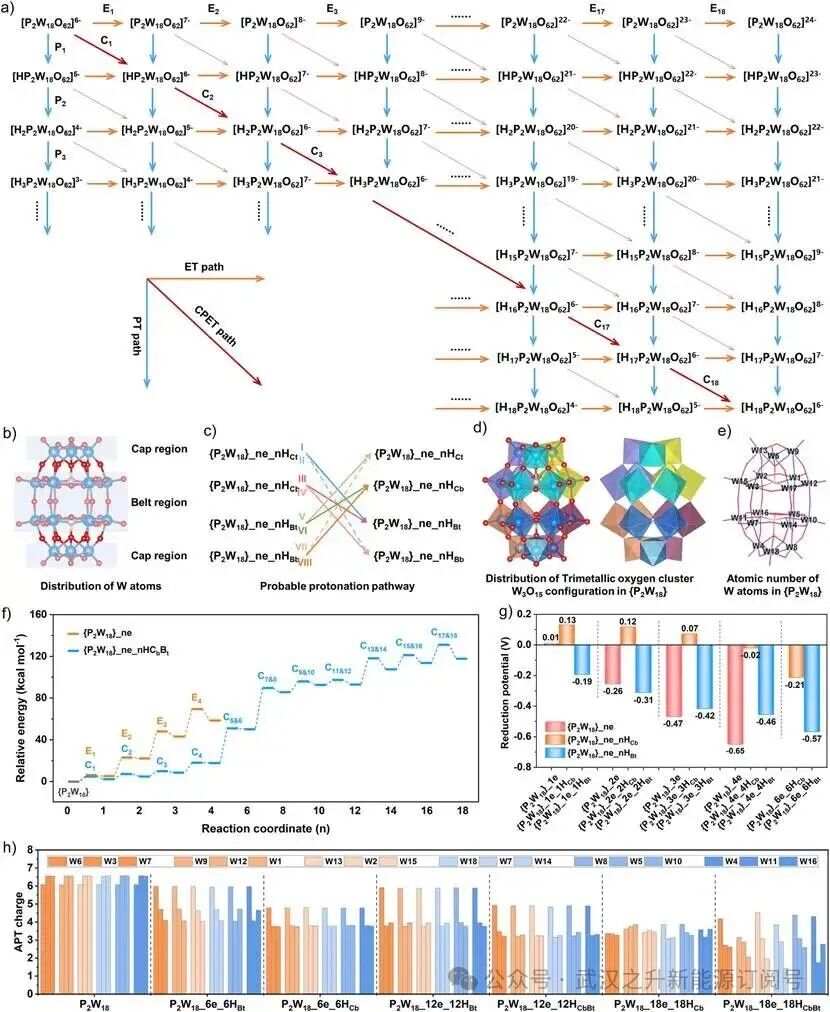
图2. {P2W18}氧化还原过程的质子耦合电子转移理论分析。(a){P₂W₁₈}的PCET过程示意图。(b){P₂W₁₈}簇中W原子的区域分布。(c)PCET路径中{P₂W₁₈}还原过程的可能质子化路径。(d){P₂W₁₈}中三金属氧簇(W₃O₁₅)构型的分布。(e){P₂W₁₈}中W的原子编号。(f){P₂W₁₈}在ET路径中还原过程的活化能网络图,以及{P₂W₁₈}_ne_nHCbBt在CPET路径中的活化能网络图。(g)低还原态(n=1-6)下{P₂W₁₈}_ne_nHCb和{P₂W₁₈}_ne_nHBt的还原电位。({P₂W₁₈}_ne_nHCb:{P₂W₁₈}簇“帽区”的桥氧质子化;{P₂W₁₈}_ne_nHBt:{P₂W₁₈}簇“带区”的端氧质子化)(h){P₂W₁₈}_ne_nH(n=0, 6, 12, 18)在三金属氧簇中各W位点的原子极化张量(APT)电荷。
{P2W18}复杂的18电子转移过程通过质子耦合电子转移(PCET)机制进行,其反应网络如图2a所示,主要包括三种主导路径:协同质子–电子转移(CPET)、顺序质子–电子转移(SPET)和顺序电子–质子转移(SEPT)。高度还原但未质子化的物种会受到静电排斥限制,而过度质子化但未还原的物种则会受质子饱和限制,二者均可能导致{P2W18}团簇结构的不可逆变形甚至坍塌。因此,作者重点关注{P2W18}在还原和质子化过程中的CPET(简写为C)、ET(电子转移,简写为E)和PT(质子转移,简写为P)路径。
基于已识别的{P2W18}团簇在“帽区”和“带区”中桥氧和端氧上的质子化热点(图2b、图S1d及图S6-S9),通过计算模拟了低还原态下{P2W18}_ne的ET构型及{P2W18}_ne_nH(n≤6)的CPET构型,以及高还原态下{P2W18}_ne_nH(6<n≤18)的CPET构型。如图2c和图S10所示,考虑到高还原态下{P2W18}“帽区”和“带区”中Oₜ和Ob的质子化,CPET过程中存在8条可能的质子化路径,分别对应于{P2W18}_ne_nHCBt、{P2W18}_ne_nHBCt、{P2W18}_ne_nHCBb、{P2W18}_ne_nHBCb、{P2W18}_ne_nHCbBt、{P2W18}_ne_nHBtCb、{P2W18}_ne_nHBbCt和{P2W18}_ne_nHCtBb。然而,通过图S14的热力学统计分析,{P2W18}_ne_nHBbCt和{P2W18}_ne_nHCtBb两条路径被排除。吉布斯自由能分析确定{P2W18}_ne_nHBt和{P2W18}_ne_nHCb为热力学上更有利的CPET路径(图S11-S12)。因此,作者通过Marcus理论和密度泛函理论(DFT)计算,系统研究了{P2W18}_ne_nHCBb和{P2W18}_ne_nHBCt路径经CPET过程的活化能(ΔG≠)网络图(图S15)。这些结果表明,CPET路径中的ΔG≠呈现两阶段演化特征:{P2W18}_ne_nHCBb路径主导低还原度(1-6e⁻)过程,而{P2W18}_ne_nHBCt路径在高还原度(>6e⁻)过程中占优。在此基础上,系统研究了Ob和Oₜ位点同时质子化的混合质子化体系{P2W18}_ne_nHCbBt。相应的CPET路径表现出比单一位点质子化体系更低的ΔG≠,这是因为相关的电子重构稳定了还原态构型,从而促进了持续的多电子存储。此外,如图2f所示,在{P2W18}相同还原度下,CPET路径的ΔG≠低于分步进行的ET过程,表明当{P2W18}发生质子耦合电子转移反应时,CPET路径在动力学上更为有利。
为了研究{P2W18}及其还原产物的氧化还原性质,作者在图2g中计算了ET和CPET路径下的还原电位。{P2W18}_1e、{P2W18}_2e、{P2W18}_3e和{P2W18}_4e的还原电位分别为0.01 V、-0.26 V、-0.47 V和-0.65 V(vs. SHE)。然而,与实验CV数据相比,{P2W18}_ne计算还原电位明显的负移凸显了质子化在{P2W18}还原过程中的关键作用。低还原态下{P2W18}_ne_nHCb物种的还原电位分别为0.13 V、0.12 V、0.07 V、-0.02 V和-0.21 V(vs. SHE),与图1b中的实验CV数据高度吻合。此外,{P2W18}_ne_nHBt物种的计算还原电位存在显著偏差,而{P2W18}_ne_nHCbBt物种在所有还原态下均表现出更合理的还原电位(表S8)。这些结果证实,Ob质子化和Oₜ质子化分别通过降低低还原态和高还原态下{P2W18}_ne_nH还原物种的还原电位,主导了CPET路径。
如图2d所示,{P2W18}团簇具有六个用不同颜色标记的三金属氧簇单元(W3O15),每个单元包含一个“帽区”W原子和两个通过桥氧原子连接的“带区”W原子。{P2W18}团簇中18个W位置的原子编号见图2e。通过分析所有活性位点的电荷发现,在CPET路径中W和O原子的原子极化张量(APT)电荷持续降低。图2h直观对比了{P2W18}、{P2W18}_6e_6HCb、{P2W18}_12e_12HCbBt和{P2W18}_18e_18HCbBt物种的APT电荷。{P2W18}团簇中所有W原子呈现均质的APT电荷(平均+6.4),其中帽区W(+6.1)的电荷密度低于带区W(+6.6),表明由区域配位不对称性导致的本征电子梯度。然而,对于{P2W18}_6e_6HBt、{P2W18}_6e_6HCb、{P2W18}_12e_12HBt和{P2W18}_12e_12HCbBt簇,帽区W原子的APT电荷均高于带区W原子,其值分别为5.96对4.37、4.79对3.78、5.88对3.85、4.89对3.28,证明电子优先存储在“带区”以实现CPET路径中W原子的还原。此外,{P2W18}_18e_18HCb和{P2W18}_18e_18HCbBt簇实现了每个三金属氧簇单元内W原子的电荷均质化,证实了{P2W18}团簇中W原子的区域选择性还原顺序遵循“从带区到帽区”。
总体而言,电子优先聚集在“带区”W位点,质子化则随还原程度加深从低还原度下的桥氧(Ob)转向高还原度下的端氧(Ot),从而形成一种低能垒、区域选择性的CPET路径,稳定了高还原态的{P2W18}团簇。
3. {P2W18}不同充电状态的结构演化
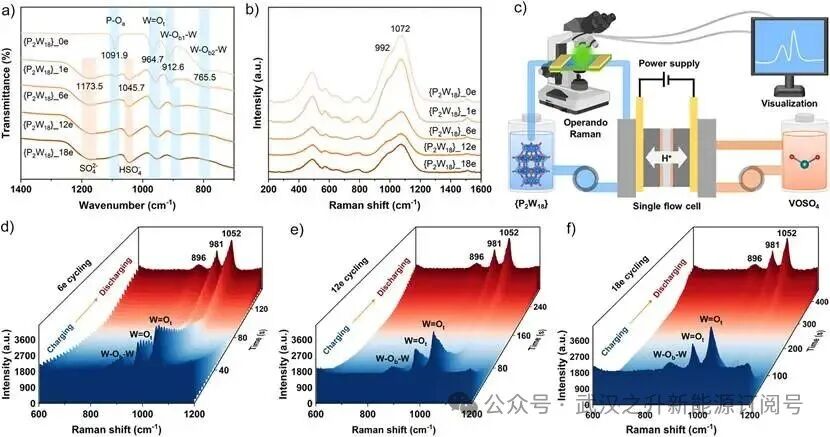
图3. {P2W18}不同充电状态的结构演化。(a)还原态{P₂W₁₈}_ne(n = 0, 1, 6, 12, 18)的傅里叶变换红外(FTIR)光谱。(b)还原态{P₂W₁₈}_ne(n = 0, 1, 6, 12, 18)的拉曼光谱。(c)液流电池原位拉曼测试示意图。H₆{P₂W₁₈}-钒液流电池(HPVBs)在(d)6e⁻容量循环、(e)12e⁻容量循环和(f)18e⁻容量循环期间的工况流动拉曼光谱3D图。
为阐明{P2W18}电解液在深度还原过程中的结构演化,使用FTIR和Raman光谱揭示了{P2W18}_ne电解液在不同还原态(1e⁻, 6e⁻, 12e⁻, 18e⁻,以原始氧化态0e⁻为参照)下的结构响应。如图4a所示,位于1091.9 cm⁻¹、964.7 cm⁻¹、912.6 cm⁻¹和765.5 cm⁻¹的FTIR特征峰分别归属于P–Oa对称伸缩振动、W=Od不对称伸缩振动、W–Ob–W不对称伸缩振动和W–Oc–W弯曲振动,与{P2W18}粉末材料的FTIR结果高度一致(图S2a-b)。此外,在1173.5 cm⁻¹和1045.7 cm⁻¹处新观察到的特征峰分别归属于SO42⁻和HSO4⁻中S–O键的不对称伸缩振动信号,其在原始{P2W18}电解液中未观察到,这与{P2W18}_ne电解液中稀释的H2SO4介质有关。值得注意的是,随着SoC增加,FTIR和Raman特征峰强度逐渐减弱,这是由{P2W18}簇的质子化和电荷重排引起的(图4a-b)。显著地,{P2W18}_ne电解液相较于固体样品更宽的拉曼峰(图S2c-d)源于溶剂化环境和分子运动状态的差异(图4b),其中溶剂化壳层效应促进了1072 cm⁻¹(W=Ot对称伸缩振动)和992 cm⁻¹(W=Ot不对称伸缩振动)峰之间的重叠。
进一步地,采用工况流动拉曼光谱(示意图见图4c)追踪了HPVB运行期间{P2W18}在可逆CPET过程中的氧化还原化学和结构演化。HPVB的充放电过程分别对应于H6{P2W18}负极电解液的还原和氧化。如图4d-f中3D图所示,1052 cm⁻¹处的W=Ot对称伸缩振动、981 cm⁻¹处的W=Ot不对称伸缩振动和896 cm⁻¹处的W–Ob–W伸缩振动的强度,在充电至{P2W18}预设容量(6e⁻, 12e⁻和18e⁻)过程中逐渐减弱,随后在放电过程中恢复。值得注意的是,随着{P2W18}充电深度增加(6e⁻→12e⁻→18e⁻容量),W=Ot和W–Ob–W键的振动强度进一步减弱。此外,与质子化O位点相关的W=Ot和W–Ob键长随{P2W18}团簇的CPET进程呈现规律性增长(表S9-S14),表明{P2W18}团簇的电子重构和质子化诱导了中间体W–O–H键的弛豫,并削弱了原始键的伸缩振动强度。
4. {P2W18}多电子转移潜能及氧化还原控制步骤
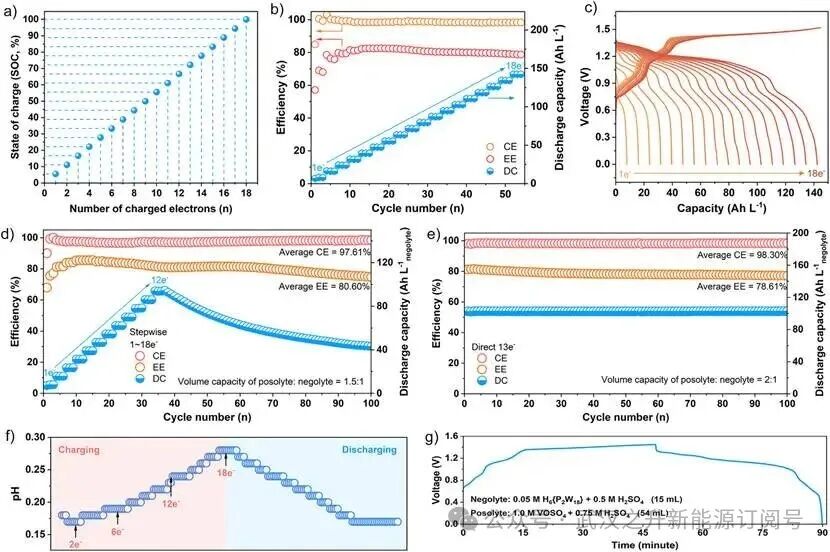
图4:{P2W18}多电子转移潜能及氧化还原控制步骤分析。(a)还原态{P₂W₁₈}的SoC定义。(b)基于{P₂W₁₈}的RFB的逐步充放电性能。负极液:0.3 M {P₂W₁₈} + 1.8 M H₂SO₄;正极液:1.5 M VOSO₄ + 3.0 M H₂SO₄;正极液的理论体积容量设置为负极液的三倍。(c)逐步充放电过程中的恒电流电压–容量曲线。(d)在有限质子供应条件下{P₂W₁₈}氧化还原过程中控制步骤的监测。(e)在充足质子供应条件下{P₂W₁₈}氧化还原过程中控制步骤的突破。(f)HPVB充放电过程中的原位pH值变化。(g)原位pH监测循环过程中的电压–时间曲线。
为建立标准化分析框架,将{P2W18}的完全还原态(18e⁻转移)定义为100%充电状态(SoC),对应于理论电子存储容量。如图4a所示,SoC百分比与电子转移数(n)保持严格的线性比例关系(SoC=5.56n%),表明每个转移电子贡献约5.56%的SoC。通过设置24电子容量和1.75 V电压的充电截止限,使用0.2 M H6{P2W18}负极电解液研究了HPVB的过充行为。如图S23所示,充电过程在达到24电子容量前触及电压截止,随后的放电过程仅能实现11.7电子容量,库仑效率为57.9%,这与之前的报道一致。证实了在RFB系统运行窗口内超过定义{P2W18}的100% SoC是不可行的,因此建立容量截止条件是防止过充的安全边界。通过逐步充放电循环,组装了以H6{P2W18}为负极、VOSO4为正极的HPVB,在不同电子容量下的三周期运行中展示了稳定的多电子(1至18e⁻)转移循环性能(图4b)。在100 mA cm⁻2电流密度下,放电容量(DC)经过54个循环(超过90.8小时)逐步增加至141.87 Ah L⁻¹,相当于0.3 M H6{P2W18}负极电解液100% SoC,平均库仑效率(CE)为98.5%,能量效率(EE)为79.6%。此外,HPVB充放电循环过程的电压–容量曲线中出现了三个明显的平台区域(图4c)。充电过程显示三个平台,对应于依次储存1-2e⁻、3-4e⁻和5-18e⁻电子容量的步骤;而放电过程中的三个平台则对应于依次释放1-2e⁻、3-14e⁻和15-18e⁻电子容量的步骤。这些发现最终证明了{P2W18}团簇卓越的多电子氧化还原能力,通过逐步循环解锁了完整的18e⁻转移,这是超越传统POM基储能材料的关键进展。然而,HPVB在高SoC下的长期稳定可逆循环性能仍需系统研究。
{P2W18}还原的CPET路径涉及多个电子和质子的参与,具体由18个串联的单电子转移步骤组成。根据实验,这些连续单电子转移步骤的动态行为表现出异质性,由于{P2W18}团簇内部的电子重构或外部质子迁移,某些步骤通常涉及一个或多个控制步骤(速率决定步骤)。结合电化学测试和RFB的性能相关性,作者假设在{P2W18}的CPET路径中,第7个(控制步骤I)和第13个(控制步骤II)电子转移步骤可能存在动力学控制(图S20)。在玻碳电极(活性面积0.07 cm2)作为工作电极的三电极配置下,{P2W18}电解液的CV测试显示出约6e⁻转移(33.3% SoC)。这种更多电子转移的限制源于严重的浓差极化(静态电解液)和有限的活性位点,导致只能检测到控制步骤I内的电子转移。然而,在恒电流充放电和强制电解液对流条件下,{P2W18}电解液在RFB系统的阴极(碳毡,活性面积9 cm²)可以持续获得稳定的电子供给和释放,从而有可能突破控制步骤I并实现更多电子转移(>6e⁻)。此外,值得注意的是,在HPVB运行期间,{P2W18}团簇在负极发生可逆的氧化还原反应。充电时,{P2W18}被还原,同时质子(H⁺)从正极通过膜迁移以维持电荷中性,从而完成电流回路。放电时,{P2W18}被氧化并伴随去质子化,质子反向扩散回正极。该模型促进了充电期间通过CPET路径对{P2W18}的还原,并使得放电期间{P2W18}的氧化耦合去质子化的高效进行。
更重要的是,当质子供应不足时,{P2W18}还原的控制步骤II(13e⁻步骤)将在RFB系统中显现,例如在基础配置中(负极液:0.3 M H6{P2W18} + 1.8 M H2SO4, 8 mL;正极液:1.5 M VOSO4 + 3 M H2SO4, 43.2 mL;容量比1.5:1)。随后,对HPVB开展从1e⁻到18e⁻的逐电子容量测试,在前36个循环(1e⁻到12e⁻步骤)中表现出稳定运行,最大DC达到93.78 Ah L⁻¹。然而,从13e⁻步骤开始,充电电压极化的急剧增加引发了DC的逐渐衰减,表明由于“质子饥饿”,{P2W18}团簇处于还原电位升高的不稳定状态。100次循环后,DC最终衰减至43.14 Ah L⁻¹(图4d)。值得注意的是,调整理论容量比(正极液容量:负极液容量=3:1)后,图4b中原本在控制步骤II观察到的充电电压极化消失了。此外,HPVB在13e⁻容量下直接充放电运行100个循环表现出显著的稳定性,DC保持在102.83 Ah L⁻¹无衰减(图4e),平均CE和EE分别为98.3%和78.61%。如图4f-g所示,通过原位pH系统(图S21)监测了H6{P2W18}负极液在充放电过程中的pH演变,其在HPVB运行期间表现出不同的pH区间。在初始2e⁻容量充电过程中,pH从0.18降至0.17并稳定,表明H⁺在{P2W18}的溶剂化壳层中积累,可能尚未发生有效的质子化。对于3e⁻至18e⁻的充电过程,pH从0.17单调增加至0.26,证明跨膜H⁺不足以质子耦合还原过程且需要从负极液中额外提取质子。相比之下,在放电阶段,pH从0.26降至0.17,证实了质子从{P2W18}团簇脱质子化并通过跨膜传输重新进入正极液。
5.HPVB液流电池性能测试
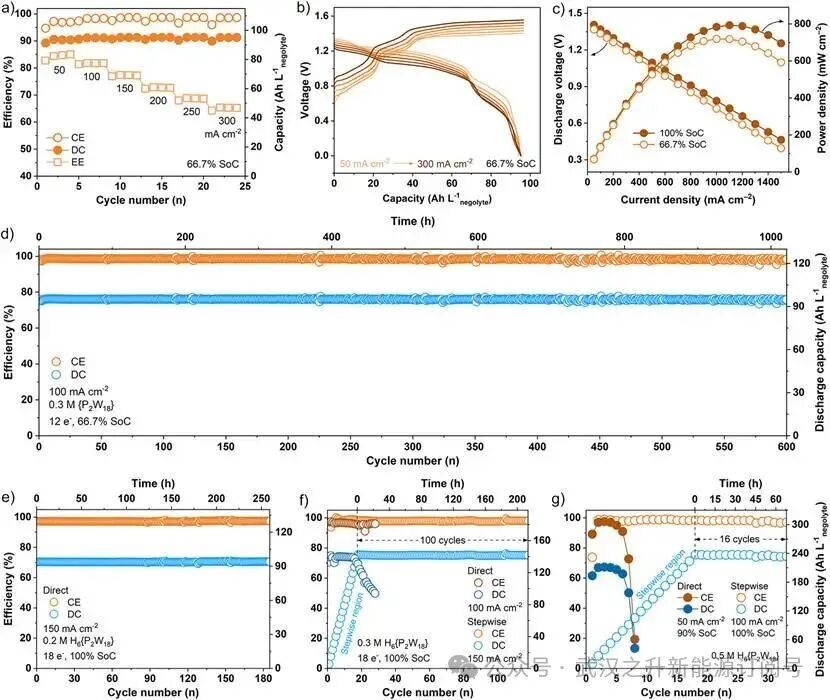
图5. HPVB液流电池性能分析。(a)HPVB单电池在0.3 M H6{P2W18}负极液66.7% SoC下,电流密度从50 mA cm⁻²至300 mA cm⁻²循环时的容量和效率。(b)在0.3 M H6{P2W18}负极液66.7% SoC下,从50 mA cm⁻²到300 mA cm⁻²的恒电流电压–容量曲线。(c)0.3 M H6{P2W18}负极液在66.7% SoC和100% SoC下的放电极化曲线。(d)在100 mA cm⁻²电流密度和0.3 M H6{P2W18}负极液66.7% SoC下的放电容量(DC)和库仑效率(CE)。(e)0.2 M H₆{P₂W₁₈}负极液在100% SoC下通过直接充放电协议的DC和CE。(f)0.3 M H6{P2W18}负极液在100% SoC下通过直接和逐步充放电协议的DC和CE。(g)0.5 M H6{P2W18}负极液在100% SoC下通过直接和逐步充放电协议的DC和CE。
基于对{P2W18}团簇CPET机理的深入理解,作者设计了高质子活性的H6{P2W18}负极液和用于HPVB的逐步充放电协议,以克服{P2W18}在高SoC下长期存在的“质子饥饿”瓶颈(表S16)。因此,通过直接和逐步充放电协议,系统评估了HPVB单电池(图S16和S17)在{P2W18}高SoC(66.7% SoC(12e⁻)和100% SoC(18e⁻))下的循环性能。HPVB单电池采用0.3 M H6{P2W18}于1.8 M H2SO4作为负极液,与1.5 M VOSO4于3 M H2SO4的正极液配对,确保{P2W18}氧化还原过程的CPET路径中有充足的质子协同作用。在酸性条件下,VO2⁺/VO2⁺氧化还原电对在0.991 V vs. SHE处表现出高度可逆的单电子氧化还原反应。在0.3 M H6{P2W18}负极液66.7% SoC下,HPVB在50至300 mA cm⁻²电流密度范围内表现出优异的倍率性能(图5a,b)。随着电流密度增加,充放电平台之间的电压极化逐渐增大,EE从85.05%系统性下降至65.37%。此外,在0.3 M H6{P2W18}负极液66.7% SoC和100% SoC下分别实现了719.2 mW cm⁻²和793.22 mW cm⁻²的高功率密度(图5c)。进一步地,如图5d所示,采用直接充放电协议的HPVB单电池在100 mA cm⁻²电流密度和0.3 M H6{P2W18}负极液66.7% SoC下表现出卓越的循环稳定性,经过600次循环(超过1020小时)放电容量无明显衰减,期间平均DC为95.04 Ah L⁻¹,CE为98.50%。
更为显著的是,采用直接充放电协议的HPVB单电池在0.2 M H6{P2W18}负极液100% SoC下运行180个循环(超过250小时),平均DC为93.84 Ah L⁻¹,CE为97.27%(图5e)。然而,当在直接充放电协议下运行时,HPVB单电池在0.3 M H6{P2W18}负极液100% SoC下经历了快速的容量衰减(图5f)。该结果归因于充电过程中质子供应相对于高需求不足,随后触发了截止电压。电子的过度积累可能导致WO6八面体畸变,造成部分{P2W18}团簇失活,并在持续循环过程中以容量衰减的形式导致电池失效。此外,作者为HPVB单电池设计了逐步充放电协议,以确保H6{P2W18}负极液在100% SoC下的稳定运行,克服因高浓度H6{P2W18}在酸性介质中的溶解度有限而导致的初始质子匮乏条件(图5f-g)。令人鼓舞的是,0.3 M H6{P2W18}负极液在150 mA cm⁻²电流密度下表现出优异的DC(141.75 Ah L⁻¹)和能量密度(ED,137.13 Wh L⁻¹),CE为97.95%(图5f)。最重要的是,如图5f所示,0.5 M H6{P2W18}负极液在100 mA cm⁻2电流密度和100% SoC下实现了破纪录的DC(236.03 Ah L⁻¹)和ED(239.02 Wh L⁻¹),CE为97.86%(表S17)。原位pH结果表明,放电阶段H6{P2W18}可能存在轻微的不完全去质子化,这反过来通过累积效应促进了H6{P2W18}负极液在100% SoC下的循环。因此,逐步充放电协议有助于降低{P2W18}在高SoC下的高质子能垒,突破“质子饥饿”瓶颈,有效提高了基于{P2W18}的RFB的循环稳定性。如表S18和S19总结,本工作开发的H6{P2W18}负极液提供了卓越的性能指标。最值得注意的是,其放电容量和能量密度显著超过了典型的POM-RFB(通常DC < 80 Ah L⁻¹)以及许多最先进的RFB系统,例如全钒(ED < 40 Wh L⁻¹)、锌溴(ED ≤ 152 Wh L⁻¹)、先进有机(ED < 60 Wh L⁻¹)和氧化还原靶向(液-固,ED ≤ 97.4 Wh L⁻¹)RFB,这确立了H6{P2W18}作为更高能量密度RFB应用的理想POM候选者。FTIR、拉曼以及高分辨质谱(HRMS)结果证实,{P2W18}电解液在循环前后的结构保持稳定(图S24和S25)。此外,高质子活性的H6{P2W18}负极液在不同浓度下均表现出高电导率(>436 mS cm⁻¹,表S20),支持了{P2W18}还原过程中的CPET过程。还需提及的是,基于{P2W18}的CPET机制,质子在活性位点被有效消耗,从而在一定程度上抑制了HPVB循环期间可用于析氢反应的局部质子可用性。虽然粘度随SoC增加而上升,但0.5 M H6{P2W18}负极液在100% SoC时的最高粘度(25 °C下82.52 mPa·s,表S20)与已报道的0.5 M H6{P2W18}负极液在33.3% SoC时的粘度以及1.5 M AQ-1,8-3E-OH负极液的粘度相近。为了向实际应用推进,未来的工作需要通过专门的电解液工程设计,重点缓解H6{P2W18}负极液在高浓度下质子供应不足和在高SoC下粘度上升的问题。
结论展望
综上所述,该研究通过结合CPET机理阐明、高质子活性电解质工程以及稳定循环氧化还原液流电池验证的综合方法,解决了POM电解质实现高SoC操作这一长期存在的挑战。具体而言,该研究通过协同应用Marcus理论、第一性原理密度泛函理论DFT计算和从头算分子动力学AIMD模拟,证明了端氧和桥氧位点的质子化通过区域选择性CPET过程稳定了{P2W18}团簇的还原态钨位点。原位拉曼光谱与原位pH监测进一步证实了{P2W18}氧化还原过程中质子耦合的可逆性。基于这些机理见解的指导,该研究设计了高质子活性的H6{P2W18}负极电解液,并开发了逐步充放电协议,以突破{P2W18}在高SoC下稳定性和可逆性差的瓶颈。所得氧化还原液流电池在H6{P2W18}负极电解液高SoC下表现出优异的循环性能:在0.3M H6{P2W18}负极电解液66.7% SoC下采用直接充放电协议循环600圈(超过1020小时)后放电容量无明显衰减(95.04 Ah L⁻¹);在0.3M H6{P2W18}负极电解液100% SoC下采用逐步充放电协议实现了141.75 Ah L⁻1的优异放电容量;以及在0.5M H6{P2W18}负极电解液100% SoC下实现创纪录的236.03 Ah L⁻1放电容量和239.02 Wh L⁻1能量密度。这些发现将{P2W18}定位为高能量密度氧化还原液流电池中极具前景的多电子氧化还原活性物种,并通过协同质子-电子转移为解锁POM基RFBs的全SoC提供了可行范式。
HAN Mingjun, LIU Yuyang, HU Wenjihao, SUN Wei, YAN Jun, ZHOU Qiusheng, LUO Jian, GAN Lei, LIU Jianye, ZHANG Chenyang*, LI Jie*, LI Xiaobin. Unlocking Full State-of-Charge of Polyoxometalate for High-Energy-Density Redox Flow Batteries via Concerted Proton-Electron Transfer[J]. Angewandte Chemie International Edition, 2026, e18906.
https://doi.org/10.1002/anie.202518906.
延伸阅读(张晨阳&李洁团队近期论文合集):
【论文赏析】中南大学李洁综述ESM:基于多金属氧酸盐团簇的新兴氧化还原液流电池(POM-RFB):性能指标、应用前景和发展策略