
第一作者:丁文龙
通讯作者:廖俊斌 & 沈江南
通讯单位:浙江工业大学

为实现大规模、长时储能目标,全钒液流电池(VRFBs)已成为极具发展前景的电化学储能方案。这类储能系统对膜材料的稳定性与选择性提出了严苛要求。然而,传统微相分离膜在追求高离子传导率与抑制活性物质交叉渗透之间往往存在权衡效应,且长期化学稳定性不足的问题进一步加剧了这一矛盾。
为解决上述挑战,本研究,浙江工业大学沈江南/廖俊斌团队设计了一种基于超分子作用介导的策略,在离子传输通道内原位合成共价有机框架(COFs)。高磺化度的聚合物链提供磺酸基团(SO3H),作为COF前驱体的超分子作用位点,为晶体生长提供规整模板。优化后的复合膜在离子通道内形成分子筛分效应,展现出优异的离子选择性(VO2+渗透率P:3.24×10−8 cm2s−1;选择性S:5.70×105)与稳定的电池运行性能(500次循环后库仑效率CE>99.6%,能量效率EE>78.89%)。该性能优于Nafion 212膜及多种已报道的同类膜材料。
相关成果以“Self-Assembly of Covalent Organic Frameworks within Sulfonated Polysulfone Membranes via Supramolecular Interaction for Vanadium Redox Flow Batteries”为题发表在Journal of Membrane Science期刊上。
感谢浙江工业大学沈江南&廖俊斌团队
(第一作者:丁文龙博士)供稿!
本文所用
一体化液流单电池测试系统
(YTH-1/LSB-1)
及钒电解液
由武汉之升新能源有限公司提供


为解决传统离子膜固有问题,浙江工业大学沈江南/廖俊斌团队提出超分子相互作用介导策略构建具有共价有机框架离子筛的聚合物−COF复合膜。结合微相分离与COF亚纳米级孔道特性,通过尺寸排阻与Donnan效应实现高离子选择性。
本研究采用超分子相互作用介导策略,在离子传输通道内原位合成了高离子选择性COF复合膜。具体而言,我们巧妙地选用磺化聚砜作为有序模板。其磺酸根基团(−SO3−)与胺类单体间存在强超分子相互作用,使COF前驱体实现定向排列,进而促进COF在聚合物基质内发生原位“定向”或“有序”生长。磺化度为60%的磺化聚砜膜不仅为COF的原位合成提供了取向模板,还赋予材料优异的质子选择性。在酸性液流电池体系中,TpBD(NH2)离子筛通过质子结合形成带正电的铵盐以发挥Donnan effect,并结合孔径筛分作用,实现高效钒离子阻隔与质子传导。离子通道中TpBD(NH2)的存在显著提升了膜的离子选择性,为VRFBs的稳定运行提供了保障。同时,通过优化单体添加比例,成功制备出均一性良好的SPSF-TpBD-0.4复合膜。该膜在100 mA cm−2电流密度下的VRFBs系统中可稳定运行500循环以上,且始终保持优异的钒离子阻隔性能与高效质子传输性能。微相分离结构与COF离子筛的协同作用,赋予了所制备复合膜更优异的综合VRFBs应用性能。
1. 膜材料设计和表征
在传统共价有机框架(COF)合成中,单体在溶剂中的无规分散会导致低聚物无序生长与团聚。本研究开发了一种超分子作用介导的策略,通过模板组装与取向结晶调控COF在质子传输通道内的聚合过程。借助磺化聚砜(SPSF)聚合物优异的化学稳定性及其可调的−SO3−功能位点,设计了具有特殊离子传输通道的COF改性膜(SPSF-TpBD-X)。具体而言,如图1 (a)所示,SPSF主链上均匀分布的−SO3−基团与BD(NH2)单体中的–NH2基团之间存在强烈的静电相互作用,该超分子作用可诱导BD(NH2)单体在分散体系中规则排列(图1 (b))。
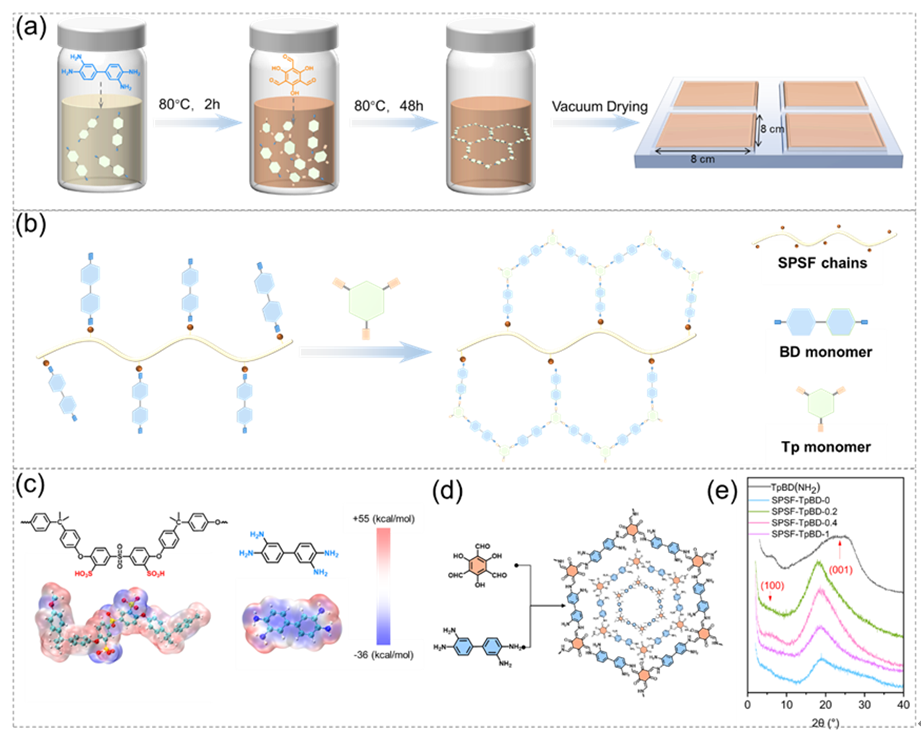
图 1. (a)共价有机框架(COF)合成路线及(b)超分子作用诱导COF合成策略示意图;(c)磺化聚砜(SPSF)链与BD (NH₂)单体的模拟静电势计算结果;(d)TpBD(NH2)的结构示意图;(e)TpBD(NH2)及SPSF-TpBD-X复合膜的X射线衍射(XRD)图谱。
通过模拟计算确定分子的静电势分布(图1 (c)),−SO3H基团呈现负静电势(–34.26 kcalmol−1),而BD(NH2)单体中的–NH2基团呈现正静电势(29.55 kcalmol−1)。由此,强酸碱相互作用促进了BD(NH2)的取向排列。随后,引入的Tp单体将优先与经诱导排列的BD(NH2)发生席夫碱反应,形成COF前驱体;继而在该半合成基底上逐步构建TpBD(NH2)共价有机框架,最终在聚合物膜的离子通道内形成均匀的COF离子筛。TpBD(NH2)的结构如图1 (d)所示。
TpBD(NH2)的XRD图谱在2θ=6.10°和25.03°处出现明显衍射峰,分别对应其(100)和(001)晶面。对于SPSF-TpBD (NH₂)-X复合膜,由于COF负载量较低,仅在2θ=6.01°处观察到微弱的COF特征衍射峰(结晶尺寸:3.63 nm)。值得注意的是,原始SPSF膜在2θ=18.10°处的衍射峰强度随COF负载量增加而逐渐减弱。
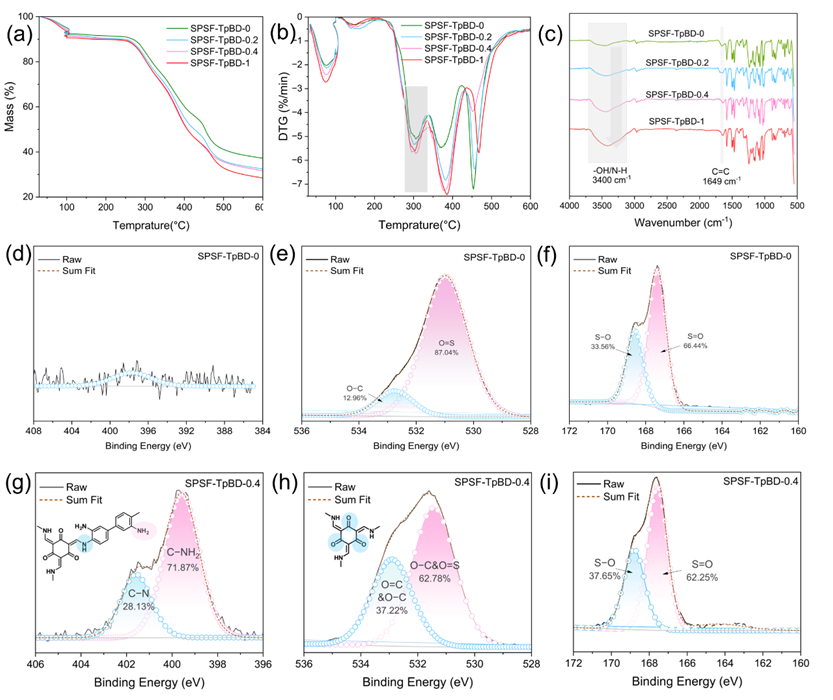
图 2. (a) 热重分析(TGA)测试曲线;(b) 微商热重分析(DTG)测试曲线;(c) 衰减全反射傅里叶变换红外光谱(ATR FT-IR);(d)、(e)、(f) 分别为 SPSF-TpBD-0 膜的高分辨 N 1s、O 1s 及 S 2p X 射线光电子能谱(XPS);(g)、(h)、(i) 分别为 SPSF-TpBD-0.4 膜的高分辨 N 1s、O 1s 及 S 2p X 射线光电子能谱(XPS)。
热重分析中,质量–温度曲线(TGA曲线)与微分热重曲线(DTG 曲线)可精准分析膜材料的热稳定性、热分解行为及组成特征。如图2 (a)和2 (b)所示,250–350℃温度区间对应侧链–SO3H基团的降解区间,此范围内降解速率随 COF 负载量增加而显著提升。该现象源于TpBD (NH₂)与–SO3−之间的超分子作用,这类相互作用削弱了磺酸基团与SPSF主链间的化学键键能,进而加速侧链降解。值得注意的是,当COF负载量达到0.4%时,侧链–SO3H基团的降解速率达到最大值;而当COF负载量进一步提升至1%时,未观察到–SO₃H基团降解速率的明显增加。此外,在主链醚键的降解温度区间(420–460℃)内,SPSF-TpBD-0.4膜表现出最慢的降解速率。由此推测,0.4%的COF负载量为最优制备条件,该推测已通过后续表征及性能测试得到验证。
图2 (c)所示的FTIR图谱表明,基底膜在1663 cm−1处的弱吸收峰在引入不同含量COF后偏移至1649 cm−1,该吸收峰归属于席夫碱反应后形成的C=C键。与此同时,SPSF-TpBD-0.4膜的高分辨N 1s光谱中,在401.6 eV处观察到C–N–仲胺键的存在,且该键与399.6 eV处C–NH2键的含量比约为1:3(图2 (g))。这与TpBD(NH2)框架单元中的化学键比例一致,为超分子作用辅助TpBD(NH2)的原位合成成功提供了直接证据。值得注意的是,引入0.4% TpBD(NH₂)后,SPSF-TpBD-0.4膜中533.1 eV处C=O峰的含量增至37.22%;531.4 eV处C–O峰的比例变化证实,Tp单体中的C–OH基团经席夫碱反应后转化为C=O基团。
2. 物理性能、钒渗透率及离子选择性
适度溶胀可促进离子在膜内快速迁移,而过度溶胀则可能降低离子交换膜(IEM)的力学性能。图3 (a)和(b)分别展示了不同膜材料在去离子水和3 molL−1硫酸溶液中的吸水率(WU)与溶胀率(SR)变化规律。随着COF单体添加量的增加,膜材料的吸水率与溶胀率整体呈现“先降低后升高”的非单调变化趋势,具体可分为两个阶段:1)当COF单体浓度为0.2%和0.4%时,TpB(NH2)有机框架在膜的离子传输通道内原位生长,生成的框架尺寸较小。这些有序排列的TpBD(NH2)框架不仅导致离子传输通道收缩,还占据部分水传输路径,最终轻微抑制膜的溶胀,实现溶胀性能优化;2)当COF单体浓度增至1%时,通道内的TpBD(NH2)有机框架出现明显团聚现象。团聚的COF颗粒破坏了通道的规整性,导致离子传输通道孔径扩大,进而使膜的溶胀率显著升高。
图3 (c)展示了所制备膜材料与Nafion 212膜的面电阻及质子传导率。经TpBD(NH2)改性后的膜电阻略有降低,其中SPSF-TpBD-0.4膜的面电阻降至0.30 Ωcm²。面电阻的降低表明质子传输阻力减小。如图3 (g)所示,该现象由离子交换模型与载体机制的协同作用导致:在离子交换模型中,酸性条件下TpBD(NH2)框架中的–NH₂基团结合质子后带正电,与相邻带负电的–SO3−基团共同为质子传导提供“跳板”,降低传导所需能量,从而实现电阻降低与传导率提升(25.57±2.02 mScm−1);另一方面,高磺化聚砜主链上离子传输通道内均匀分布的–SO3−基团形成亲水性离子簇,构建出连续的微相分离结构。连续的水通道作为质子扩散的载体,质子以水合离子形式在连续水分子间快速传导,最终实现高传导率。
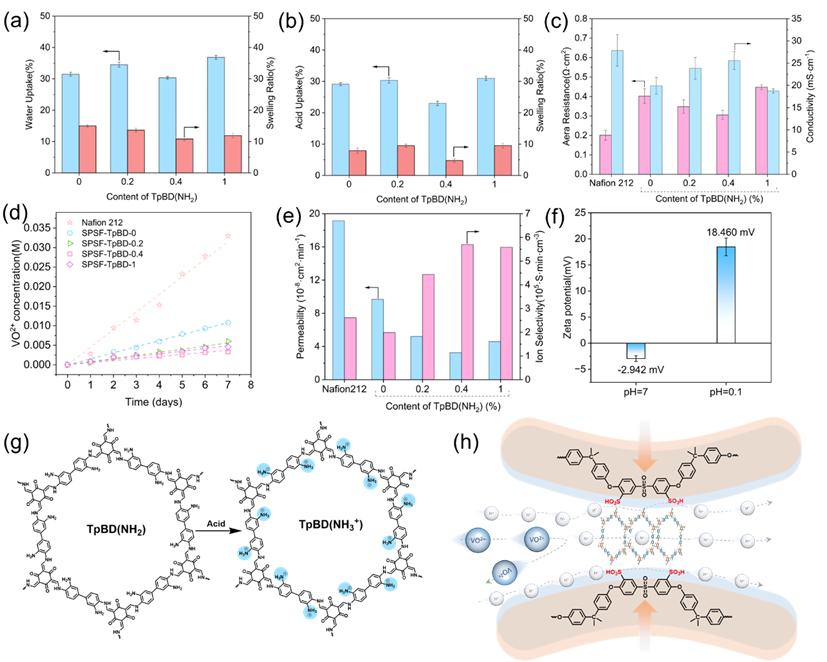
图 3. (a)吸水率(WU)与溶胀率(SU);(b)吸酸率(AU)与酸溶胀率(ASU);(c)质子传导率与面电阻;(d)扩散池接收侧VO2+浓度变化曲线;(e)钒离子渗透率与离子选择性系数;(f)酸性条件下TpBD(NH2)的zeta电位;(g)TpBD(NH2)的质子化过程示意图;(h)离子选择性作用机制示意图。
图3(d)和(e)所示,通过趋势线斜率可确定VO2+的跨膜扩散速率。基于SPSF的离子交换膜在钒离子阻隔性能上显著优于Nafion 212膜,与SPSF-TpBD-0膜相比,SPSF-TpBD-0.2、SPSF-TpBD-0.4和SPSF-TpBD-1膜的VO2+渗透率分别降至前者的1/2.34、1/3.26和1/1.80。其中,SPSF-TpBD-0.4膜的VO2+扩散速率最低(3.24×10−8 cm²s−1),远低于其他改性膜。该结果与其面电阻和质子传导率变化规律一致:TpBD(NH2)的引入填充了质子传输通道,使直径更大的水合VO2+离子在通道内扩散时受到更显著的筛分作用;同时,酸性扩散条件下,TpBD(NH2)框架中未反应的–NH₂基团结合质子后带正电,对水合VO2+离子产生显著的排斥作用(图3 (g)和(h))。随TpBD添加量增加而升高,其中SPSF-TpBD-1膜的离子选择性高达5.70×105。
Zeta电位的大小反映材料表面的电荷分布情况。如图3 (f)所示,在3.0 molL−1硫酸环境中,TpBD(NH2)表面的伯胺基团结合自由质子后带正电,这种强正电环境更有利TpBD(NH2)通过电荷间的同离子排斥作用阻挡VO2+的跨膜扩散。同时,质子化的氨基还可为质子传导提供质子交换位点(图3 (g)),显著降低质子进入通道的能垒,促进质子快速传输(图3 (h))。SPSF-TpBD-0.4膜的VO2+渗透率显著低于基底膜,为这种增强的排斥与筛分功能提供了直接证据。
在离子交换膜基质中,水合带电基团自发聚集形成亲水域,与疏水聚合物主链相互贯穿,共同构建微相分离结构,该结构被广泛认为是离子传输的“高速公路”。通过AFM相图可清晰观察到膜内亲水域与疏水域的界面特征。如图4 (a)所示,SPSF-TpBD-0膜中的亲水域呈现高度聚集状态;而随着COF的有序生长,亲水域逐渐转变为连续的长条状形貌,这一特征在SPSF-TpBD-0.4膜中最为显著。该现象可归因于COF的原位生长过程:COF占据了亲水通道内的部分空间,破坏了亲水域的聚集状态,使其从孤立团聚体转变为相互连通、均匀分布的形貌。
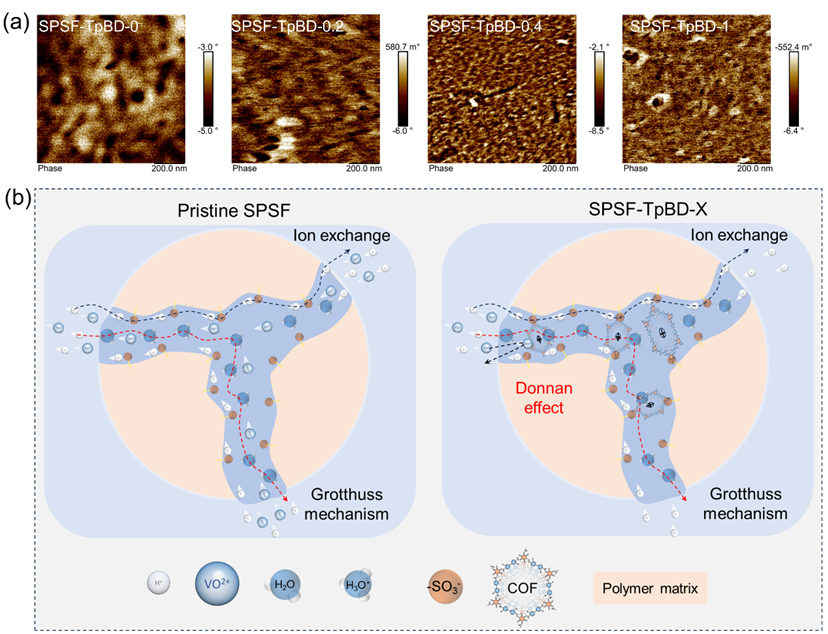
图 4.(a)SF-TpBD-X复合膜的AFM相图;(b)SPSF-TpBD-X复合膜内可能的离子选择性作用机制示意图。
图4 (b)展示了设计的搭载TpBD(NH₂)离子筛的质子传输通道,通过三种离子传输机制(离子交换机制、载体机制和Grotthuss机制)实现高选择性质子传输。在聚合物链构建的离子通道带电壁面(即富磺酸根区域)发生高速质子交换:离子突破膜表面进入传输通道后,完全水合的离子发生部分脱水并进入不稳定状态,进而与–SO3−发生相互作用;克服能垒断裂配位键后,离子跃迁至下一个跳跃位点,经多次跳跃后再次突破能垒进入渗透侧,完成跨膜离子传输。在此过程中,与VO2+相比,质子与–SO3−的相互作用更强,因此传输量更大。Grotthuss机制是实现高速、选择性质子传输的主要途径:经TpBD(NH2)改性的离子通道在酸性环境中具有更多连续水通道,质子可通过在连续相邻的水分子间跳跃实现高速传输;相比之下,四水合VO2+体积更大,通过离子筛时面临更大的空间阻力。此外,离子交换膜中固定离子交换基团的存在会形成阻碍同离子传输的屏障,即Donnan排斥效应。氨基质子化后形成的带正电铵盐增加了离子通道内的固定正电荷密度,放大了唐南电位,进一步增强了对同离子的排斥作用。
3. 单电池性能
全钒液流电池单电池测试系统是评估IEM性能最直观且关键的方法(如图5 (a)所示)。图5 (b)为SPSF-TpBD-0.4膜与Nafion 212膜的极化曲线:搭载SPSF-TpBD-0.4膜的VRFB在电流密度312.50 mAcm−2时,最大功率密度达300.03 mWcm−2;而Nafion 212膜对应的最大功率密度为284.22 mWcm−2。该结果表明,改性膜具有优异的质子传导性能,在高电流密度条件下可实现高效质子传输。图5 (c)为两种电池的开路电压(OCV)衰减曲线:在50%SOC下,SPSF-TpBD-0.4膜体系的自放电速率更慢,为Nafion膜的2.54倍。
图 5(d)为搭载SPSF-TpBD-0.4膜的电池在不同电流密度下的充放电曲线。随着电流密度升高,电池容量呈梯度下降,这归因于高欧姆极化与过电位效应。图5 (e)展示了各电流密度下5次充放电循环的测试结果:与Nafion 212膜相比,SPSF-TpBD-0.4膜电池的CE稳定在96%以上,在100 mAcm−2电流密度下经5次循环后稳定于99.3%。优异的库仑效率表明SPSF-TpBD-0.4膜具有更高的离子选择性与钒离子阻隔性能,与前文选择性测试结果一致。
搭载SPSF-TpBD-0.4膜的电池系统在能量损失控制方面表现优异:50 mAcm−2电流密度下,系统能量效率稳定于88.80%,远高于Nafion 212膜(80.1%)。随着电流密度进一步升高,所有搭载不同膜材料的电池系统均出现能量效率下降,这由高欧姆极化与过电位效应导致。在50–200 mAcm−2的测试范围内,SPSF-TpBD-0.4膜不仅效率高于Nafion 212膜,在测试稳定性方面也展现出一定优势:具体表现为5次循环的数值变化搭载 SPSF-TpBD-0.4膜的电池系统在首次循环后即达到稳定状态,而Nafion膜系统的数值在5次循环中呈持续小幅上升趋势。
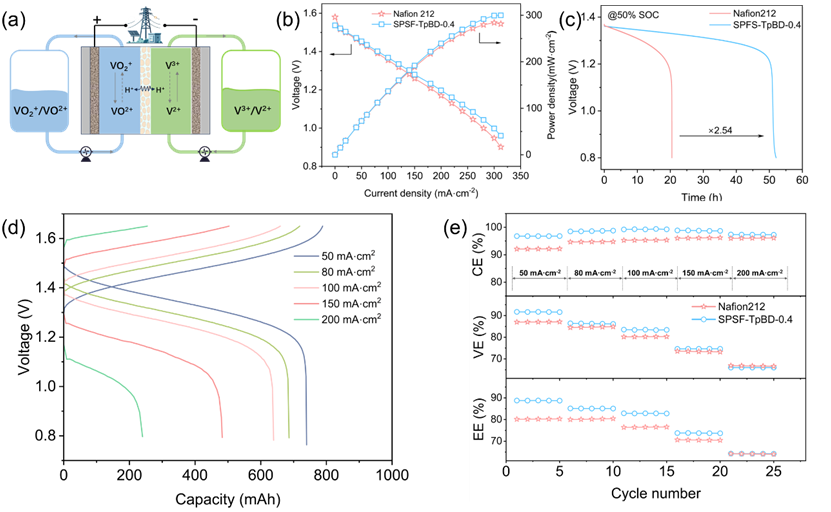
图 5. (a)全钒液流电池(VRFB)单电池装置示意图;(b)搭载SPSF-TpBD-0.4膜与Nafion 212膜的电池极化曲线;(c)50% SOC下搭载SPSF-TpBD-0.4 膜与Nafion 212膜的电池自放电曲线;(d)搭载SPSF-TpBD-0.4膜的电池在不同电流密度下的充放电曲线;(e)倍率性能图。
图6 (a)中,经500次循环后,搭载 SPSF-TpBD-0.4膜的电池放电容量保持率为 55.22%,远高于Nafion 212膜(100次循环后仅8.3%)。前者在100、300、500次循环后的容量衰减率分别为0.0712%、0.0891%、0.0895%,仅为Nafion 212膜(单圈衰减率0.91%)的五分之一。如图6 (b)所示,SPSF-TpBD-0.4膜经500次连续充放电循环后,FTIR未出现显著变化,表明隔膜基本单元结构保持完整,可耐受500次重复循环使用(总充放电时长476 h)。
图6 (c)中,长期运行后的电池在不同电流密度下的充放电曲线与图5 (d)相比虽略有偏移,但仍维持正常充放电状态。改性膜在VRFBs中的长期稳定运行,证实了经COF改性的离子传输通道具有优异化学稳定性。此外,与近期报道的钒液流电池膜材料相比,优化后的膜在容量衰减率方面展现出一定优势(图6 (d))。上述结果表明,SPSF-TpBD-0.4膜可有效阻隔钒离子渗透,且具备可靠的长期稳定性。
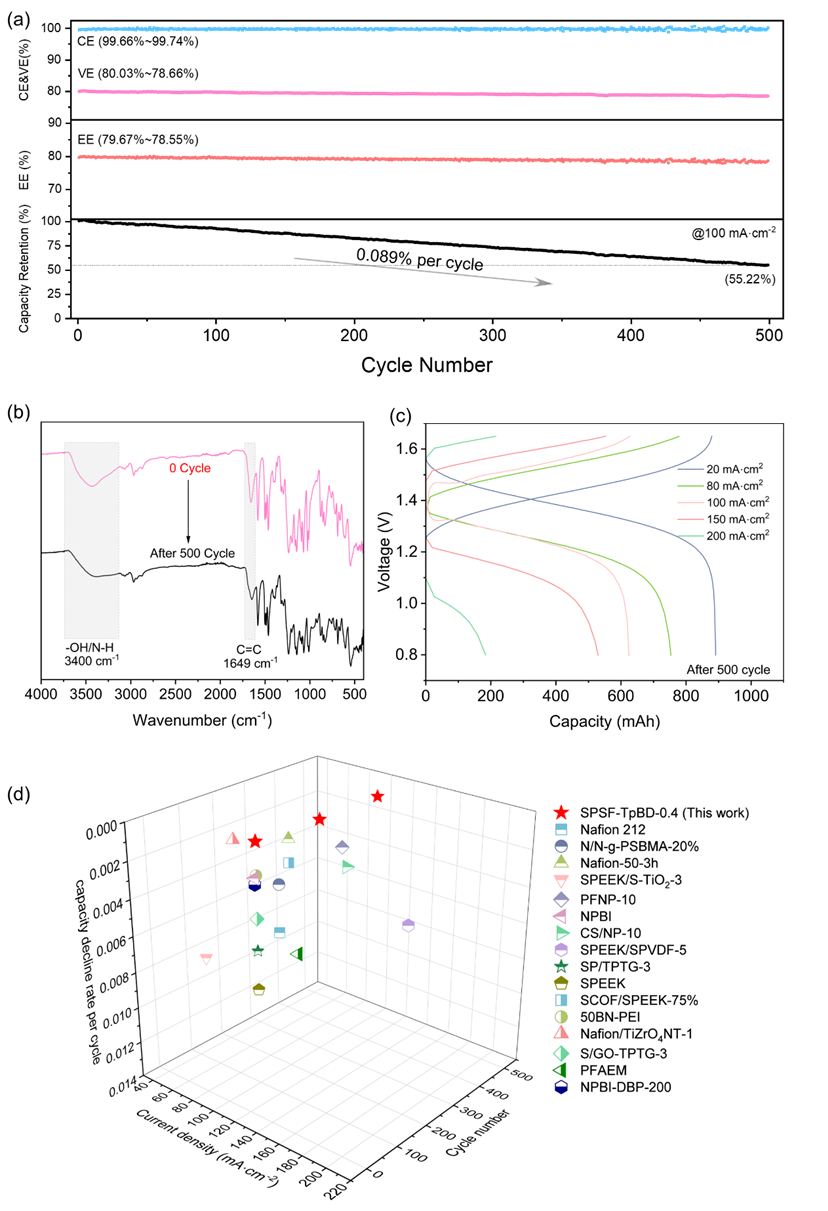
图 6. (a)搭载SPSF-TpBD-0.4膜的全钒液流电池(VRFBs)长循环测试结果;(b)SPSF-TpBD-0.4膜在 VRFBs 组装前后的傅里叶变换红外光谱(FTIR);(c)经500次循环后,搭载SPSF-TpBD-0.4膜的电池在不同电流密度下的充放电曲线;(d)不同膜材料用于VRFBs时在各电流密度下的单圈容量衰减率。
本研究成功利用超分子相互作用力辅助了聚合物内COF的原位合成。所制备的膜材料在VRFBs中表现出优异性能:在100 mAcm−2电流密度下,经500次循环后,电池系统CE保持在99.6%以上,EE稳定80%,单圈容量衰减率仅为0.089%。
Wenlong Ding, Junbin Liao, Haoyu Liu, Changrong Li, Qingsong Liu, Yanqing Xu, Jiangnan Shen, Huimin Ruan, Congjie Gao, Self-Assembly of Covalent Organic Frameworks within Sulfonated Polysulfone Membranes via Supramolecular Interaction for Vanadium Redox Flow Batteries, 2025, Journal of Membrane Science.
https://doi.org/10.1016/j.memsci.2025.124910