锌–碘(Zn-I2)液流电池以其无可置疑的安全性、可靠性、较高的理论容量、和较低的成本等优点成为一种重要的电化学储能替代技术。然而,I−/I3−氧化还原偶对的低利用效率和高可溶性聚碘化物中间体的不可控自转化仍限制着其实际应用。在此,哈尔滨工业大学张红和安徽大学鹿可团队报告了一种将FeP纳米团簇嵌入到N和P共掺杂碳框架(FeP- NPC)上构建双功能石墨毡,用于组装高能量和循环稳定的Zn-I2液流电池。通过杂原子和纳米团簇协同作用加速I−/I3−氧化还原动力学,同时极性基质与活性物质之间的强相互作用促进了界面物理化学约束效应,减轻了多碘化物的流失。因此,该Zn-I2液流电池具有优异的倍率性能,超长循环寿命达1000次,平均库仑效率达到98.1%,并大大增强了其抗自放电能力。相关成果以“A bifunctional electrocatalytic graphite felt for stable aqueous zincpolyiodide flow batteries”为题发表在国际知名期刊Journal of Power Sources上。
近年来,液流电池的广泛应用加速了相关电化学储能技术的发展。与传统电池不同,电化学液流电池通过将电活性物质储存在外部流动的电解质中,同时将氧化还原反应保持在电池内部的电极表面上,从而将功率和容量分离开来,这使得氧化还原液流电池可以独立大规模生产,同时具有很高的安全性优势。其中,锌–碘( Zn-I2 )氧化还原液流电池以其无可置疑的安全性、可靠性、较高的理论容量、和较低的成本而成为一种重要的电化学储能替代技术。特别是 I−/I3− 氧化还原偶对是最有前途的活性物质,它具有高溶解度和防止元素碘阻塞 Zn-I2 氧化还原液流系统中的泵/管道/电极的优异特性。然而, I−/I3− 氧化还原偶对的低利用效率和高溶性聚碘中间体的不可控自转化仍然是限制Zn-I2液流电池(322 Wh L−1)实际潜力的限制因素。为了应对上述挑战,许多研究人员都探索出了优化 I−/I3− 转换的可行策略。在此背景下,合理设计和修饰电极是一种可以提高吸附能力并提供电催化氧化还原反应位点以直接提高电池性能的有效途径。一般来说,在碳毡上掺杂杂原子和引入适当的电催化剂将电极功能化是实现高可逆性 Zn-I2 电池体系的常用方法。目前,N掺杂、P掺杂以及S、N共掺杂对碳材料的相关改性技术已广泛应用在Zn-I2电池体系中。例如,Lu和他同事设计了S、N共掺杂的三维石墨烯泡沫作为组装水系Zn-I2电池的电极材料,并揭示了碳基质上的S、N共掺杂杂原子可以提高基质对碘物种的捕获能力,进而催化界面碘氧化还原过程。与此类似,Zhou等人报道了一种P掺杂的多孔碳作为功能宿主,以提高Zn-I2电池内碘物种的锚定能力,并且所制备的阴极具有较高的比容量和显著的循环稳定性。将活性物种限制在掺杂杂原子的碳基质中的进展也启发了研究人员开发其他具有吸附位点和有效电催化的多功能催化材料,以提高I−/I3−转化率。Li等利用MIL-125-NH2和UiO-66-CH3等两种MOFs作为电催化基质,加速I−/I3−氧化还原反应的转化,并发现MIL-125-NH2修饰的电极具有更高的能量效率。最近,Chen和同事发现,Mo2S纳米板上的缺陷可以作为非常有效的催化位点来加速I−/I3−氧化还原反应。将原始石墨毡转化为极性催化底物是实现高效碘捕获–催化–转化过程的先决条件。金属磷化物具有增强极性和电催化能力的特点,已被广泛用于加速活性物种的氧化还原动力学。值得注意的是,磷化铁(FeP)作为一种高效的电催化剂,以其化学稳定性和低成本的优点而备受关注。且FeP 纳米团簇具有精心设计的表面特性和理想的暴露原子位点,可以更好地进入活性角位和边位,在电催化领域引起了广泛的研究兴趣。因此,FeP可以提高(聚)碘化物的化学吸附能力,同时加速I−/I3−的氧化还原动力学。然而,仅在碳毡表面引入杂原子或电催化剂,吸附和催化部位的活性往往较低。因此,当务之急是设计一种掺杂杂原子和电催化剂的双功能耦合电极,以加强对碘物质的吸收,同时实现I−/I3−的转换,并从实质上限制了I3−多碘物的泄漏和穿梭。在此,嵌入在N和P共掺杂碳框架上的FeP纳米团簇(FeP-NPC)可以构建一种双功能石墨毡,用于组装高能量和循环稳定的Zn-I2液流电池。在保持多孔石墨毡(GF)优势的同时,掺杂剂和纳米团簇协同增强(聚)碘化物的化学锚定,加速I−/I3−氧化还原动力学,以避免活性物种的穿梭和/或流失。因此,用FeP-NPC功能石墨毡组装的Zn-I2液流电池达到了在1摩尔I−和6摩尔I−的条件下分别实现了33Wh L−1和204Wh L−1的高能量密度,且库仑效率分别达到了99.4 %和99.5 %,显著媲美甚至优于采用KI阴极电解质的传统电池。此外,Zn-I2液流电池还展示了卓越的抗自放电能力和高功率密度(172 mW cm−2)。这一贡献为抑制碘穿梭和提高 Zn-I2 液流电池的能量效率提供了一种可行的方法。(1)材料的合成及结构表征
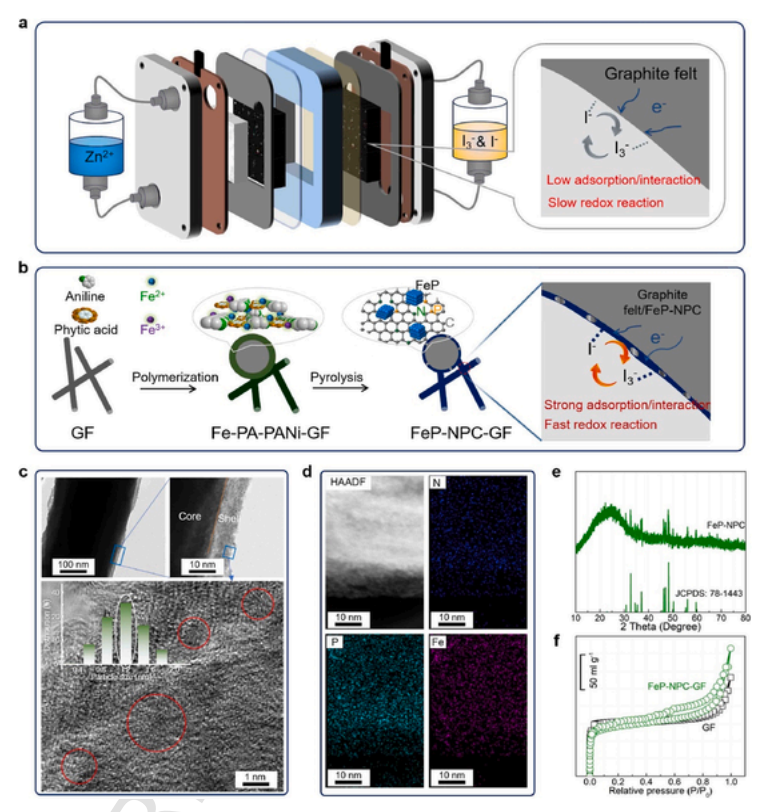
为了解决Zn-I2液流电池中活性材料与裸石墨毡相互作用弱的问题(图1a),设计了多功能石墨毡核壳结构复合材料,即将FeP纳米团簇嵌入在N和P共掺杂碳层中。图1b显示了FeP-NPC-GF复合材料的关键制备步骤,该复合材料是在原始GF上原位聚合苯胺,然后在氩气气氛下在900°C下热解1小时制成。苯胺和植酸分别作为磷和氮掺杂剂的来源,而氯化铁作为引发剂,协助FeP纳米团簇的形成。同时制备了GF上N和P共掺杂碳层(NPC-GF)的对照样品。设计这种功能性石墨毡增强了基体对I3−的吸附能力,进而实现了I−和I3−之间的高效转化。界面工程石墨毛毡的TEM图像和功能基质的相应尺寸分布如图1c所示。与未处理的GF相比,FeP-NPC-GF具有独特的核壳结构特征。形成了明显较薄的碳层,平均厚度约为10 nm,许多纳米团簇均匀地分散在碳骨架上,平均粒径分布为1.2 nm。HAADF-STEM图像和元素图谱显示了N、P和Fe元素在改性石墨毡上的均匀分布(图1d),证明了N和P在处理后的石墨毡中的成功掺杂。为了证实FeP纳米团簇的存在,我们采集了FeP-NPC-GF样品的XRD图谱和XPS光谱。X射线衍射图谱结果显示,GF的(002)和(101)衍射峰明显位于26°和44°左右,且随着热解温度的升高,峰强度略有升高,这表明石墨化程度的改善是有限的。此外,在FeP-NPC-GF样品的XRD图谱中发现了明显的衍射峰(图1e),与FeP的标准峰(JCPDS No.78-1443)很一致,证实了FeP-NPC-GF复合材料中存在FeP纳米团簇。此外,与NPC-GF的结果相比,在FeP-NPC-GF的XPS光谱中,分别出现了P 2p峰(129.3和130.1 eV)和Fe 2p峰(711.3和724.5 eV)(图1),进一步证实了FeP纳米团簇在N和P共掺杂碳框架上的成功锚定。特别地,图 1f 还得出了FeP-NPC-GF和GF的表面积,在对GF(397 m2 g-1)进行改性后,FeP-NPC-GF的比表面积(427 m2 g-1)略有增加,且观察到FeP-NPC-GF样品具有多孔结构,同时存在微孔和中孔(图2)。较薄的碳层加上较高的孔隙率有望更好地起到限制碘化物的作用。
图1。功能催化石墨毛毡的结构示意图和结构表征。(a) 重点介绍了Zn-I2液流电池的结构示意图以及阴极侧存在的问题。(b) FeP-NPC-GF复合材料的关键制造步骤。所提出的双功能催化石墨毡能够同时增强碘的物理化学限制,并引起碘的电催化氧化还原。(c) 界面工程石墨毛毡的TEM图像和相应的FeP纳米晶体的尺寸分布图像。(d) FeP-NPC-GF的 HAADF-STEM图像 和 EDS 元素图谱。(e) FeP-NPC 的 XRD 图。(f) 石墨毡和处理过的石墨毡的 N2 吸附–解吸等温线。
(2)物理化学捕获能力和电催化定向碘可逆转化
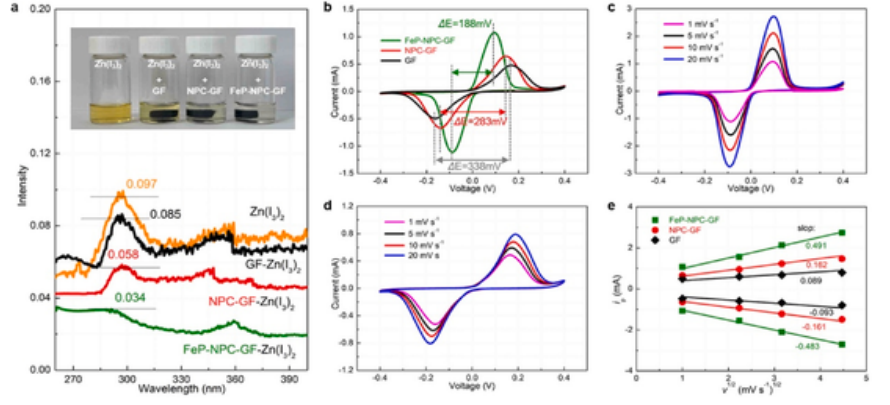
为了确定双功能基质的碘捕获性能,我们通过三碘化物捕获试验研究了宿主材料与多聚碘物之间的相互作用。紫外可见光谱中位于280和350 nm的峰归属于I3−。与新鲜Zn(I3)2溶液相比,加入GF和处理后的GF(FeP-NPC-GF和NPC-GF)后,相关吸收峰的强度减弱,这表明不同电极的不同的I3−捕获能力。如图2a所示,吸附能力从小到大依次为 GF、NPC-GF 和 FeP-NPC-GF。这种增强的限制效应可以归因于FeP-NPC-GF支架与(多聚)碘物种的化学相互作用。特别是FeP在提高FeP-NPC-GF对碘的吸收能力方面发挥着重要作用(图3),由于电荷的强相互作用和重新分布促进了类化学键的形成,这有助于功能基质实现碘物质的高效捕获–催化–转化过程。为了进一步证明基质对I3−的吸收能力,将每个阴极如GF、NPC-GF和FeP-NPC-GF分散在Zn(I3)2电解质中,保存12h。显然,与FeP-NPC-GF阴极混合的Zn(I3)2溶液的颜色变化最为明显,这表明与其他阴极相比,FePNPC-GF具有最好的吸收能力(插图,图2a)。这表明,N和P杂原子和FeP纳米团簇可以协同作用,以实现几乎完全的碘限制。进一步证明了在实际工作条件下,通过FeP和N、P杂原子的协同效应的有效捕获。 电极捕获碘物种的能力越强,其电化学性能就越好,尤其是在I−/I3−电催化氧化还原反应方面。因此,通过使用GF、NPC-GF和FeP-NPC-GF电极组装对称电池,在1 mV s-1的扫描速率下进行CV曲线测试,以验证I−/I3−氧化还原耦合的可调动力学。如图2b所示,大电流密度和小电压差表明了催化基体对改善碘氧化还原动力学具有积极作用。与NPC-GF(0.5 mA,283 mV)和GF(0.75 mA,338 mV)相比,FeP-NPC-GF的响应电流最大(1.2 mA),峰值分离最小(188 mV)。这表明FeP-NPC-GF显著增强了可逆电化学I−/I3−转化的动力学。在图2c和d中进一步比较了扫描速率从1 mV s−1增加到20 mV s−1的CV曲线。在相同的测试条件下,FeP-NPC-GF阴极的氧化还原峰比裸GF电极更窄、更尖锐,进一步证实了FeP-NPC-GF具有更强的转化动力学和较高的I−/I3−氧化还原转化可逆性。此外,所有电极的扫描速率的平方根呈线性关系,表明存在扩散受限的氧化还原反应。经典的Randles-Sevcik公式可用于评估锌离子的扩散系数。值得注意的是,FeP-NPC-GF的还原峰和氧化峰斜率均高于对照电极(图2e),这证实了理化和催化协同效应带来的最快的扩散动力学。理论结果(图4)还表明,(聚)碘物种更容易被FeP吸收,用碳中掺杂 N 和 P 也有助于进一步锚定(聚)碘,从而有效地改善可逆性能,提高循环稳定性(图4a)。此外,还对吸附的碘物质的分解过程进行了研究。图S4b表明,与NPC和石墨烯相比,碘物质在FeP表面的分解能垒大大降低,这表明FeP也能催化 Zn-I2 液流电池的充电过程。
图2。FeP-NPC-GF对碘类物质的吸附和电催化作用。(a) 在Zn(I3)2溶液中的紫外–可见吸收光谱(插图:不同样品可视化吸附试验的光学图像)。(b) FeP-NPC-GF、NPC-GF和GF对称电池在1 mV−1和1.0 M KI时的CV曲线。(c) FeP-NPC-GF和(d) GF在不同扫描速率下组装对称电池的CV曲线。(e) 不同电极的阴极峰和阳极峰与扫描速率的平方根的线性拟合,其中的斜率与离子扩散系数直接相关。
(3)碱性阴极溶液在各种温度下都很稳定
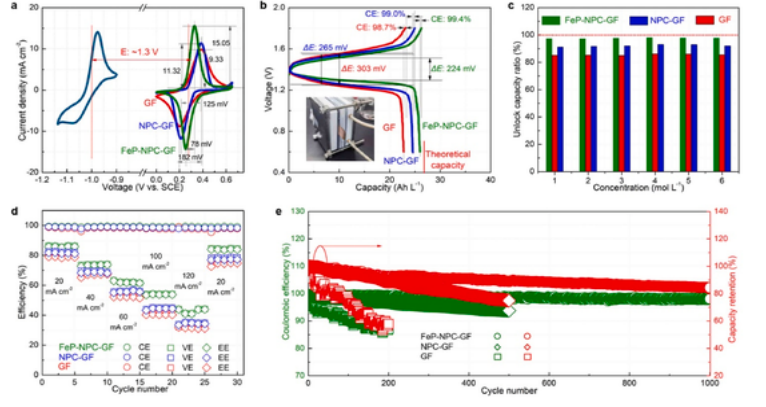
为了证明FeP-NPC-GF在电池体系中的电催化活性,我们分别对阴极和阳极进行了 CV 曲线分析,以确定了Zn2+/Zn和I−/I3−氧化还原偶的电位间隙(图3a)。典型的标准 CV 曲线在整个电压扫描范围内都没有发生氢和氧的析出反应,这意味着具有很高的可逆性,副反应可以忽略不计。在Zn-I2液流电池中FeP-NPC-GF电极的平衡电位差约为1.3V。与NPC-GF(11.32 mA cm-2,125 mV)和 GF(9.33 mA cm-2, 182 mV) 相比,FeP-NPC-GF 提供了更高的响应电流密度 (15.05 mA cm-2) 和最小的峰值分离 (78 mV),证实了 FeP 对 I−/I3− 氧化还原反应的预期电催化效果。如图 3b 所示,用不同的阴极组装了 Zn-I2 液流电池夹具(型号:LSB-1,生产厂家:武汉之升新能源有限公司),如图3b所示。与NPC-GF(265 mV)和GF电极(303 mV)相比,FeP-NPC-GF提供了更窄的峰分离(224 mV)和更高的充放电能力。在20 mA cm−2的电流密度下,FeP-NPC-GF 成功获得了 25.38 Ah L-1 的高实际容量,接近理论值。此外,基于FeP-NPC-GF的Zn-I2液流电池显示出卓越的稳定性,库仑效率(CE)高达99.4 %,而NPC-GF和GF的电极分别只有99.0 %和98.7 %。为了进一步研究FeP-NPC-GF释放I3−容量的能力,我们在Zn-I2液流电池中测试了1.0M~6.0M范围内的不同浓度的KI,见图3c。FeP-NPC-GF 组装的 Zn-I2 液流电池可以充分利用电解质中的 I−,在 1.0 M KI 浓度下具有超高容量,解锁率约为 97.7%,而基于 NPC-GF 和 GF 的液流电池容量则接近 92% 和 85.5%。即使在高 KI 浓度(6.0 M)下,FeP-NPC-GF、NPC-GF 和 GF 液流电池的相关解锁容量比略有变化,分别为 97.2 %、91.1 % 和 85.1 %。这是因为 FeP-NPC-GF 电极可以稳定 I3−,从而提高 Zn-I2 液流电池的容量。上述结果进一步证明了我们的预期,即独特的结构特征加强了对 I3− 的吸附并抑制了其流失。此外,加速了I−/I3−的转换,实现了Zn-I2液流电池优异的能量效率。图3d为FeP-NPC-GF组装的Zn-I2液流电池在不同电流密度下的CE保持稳定在99 %左右。随着电流密度的增加,能量效率(EE)和电压效率(VE)均有所下降,这是由于在高电流密度下器件存在严重的浓差极化和欧姆极化现象。因此,当电流密度从20 mA cm−2增加到120 mA cm−2时,基于FeP-NPCGF的Zn-I2液流电池的CE、EE和VE均高于NPC-GF和GF。此外,当以FeP-NPC-GF为电极时,电流密度恢复到20 mA cm−2后,EE和VE可以恢复到原始值。在电流密度为20 mA cm−2时,Zn-I2液流电池的最大能量效率为87 %。随后,进一步研究了Zn-I2液流电池的循环性能。含有6.0 M KI的FeP-NPC-GF可实现 1000 次左右的稳定充放电循环,并具有高容量保持率(95.2%)和高 CE(平均 90.3%)。而500次循环后NPC-GF的CE为86.1 %,容量保留率为91.9 %,最严重的是GF在200次循环后CE只有72.2 %,容量保留率仅为87.1 %。如图所示,随着循环测试次数的增加,放电容量和 CE 值均有所下降,而电化学响应的下降可归因于不可控的副反应、非活性碘物种的聚集和多碘化物的穿梭。如图5 所示,即使在 60 mA cm-2 的高电流密度下,FeP-NPC-GF 仍能稳定地充放电约 1200 次,容量保持率为 88.1%,平均 CE 为 97.9%。该电池良好的能量效率证明了基于FeP-NPC-GF的Zn-I2液流电池的稳定性和可逆性,这得益于I−/I3−的快速转化,进一步证实了杂原子和电催化剂在改性碳基质中的协同电催化作用。
图3。Zn-I2液流电池的电化学性能。(a) 在10 mV s−1 下,正电解质(1 M KI + 1 M KBr)在三电极电池中FeP-NPC-GF、NPC-GF和GF修饰工作电极的CV图。以及1M硫酸锌电解液中锌电极的CV曲线。(b) 在20 mA cm−2时,Zn-I2液流电池的充放电曲线。插图是组装好的液流电池夹具的实物图。(c) 不同浓度KI下,Zn-I2液流电池的容量解锁率。(d)Zn-I2液流电池在20~120 mA cm−2不同电流密度下的能量效率。(e)Zn-I2液流电池在20 mA cm−2条件下的循环性能。
(4)抗自放电性能
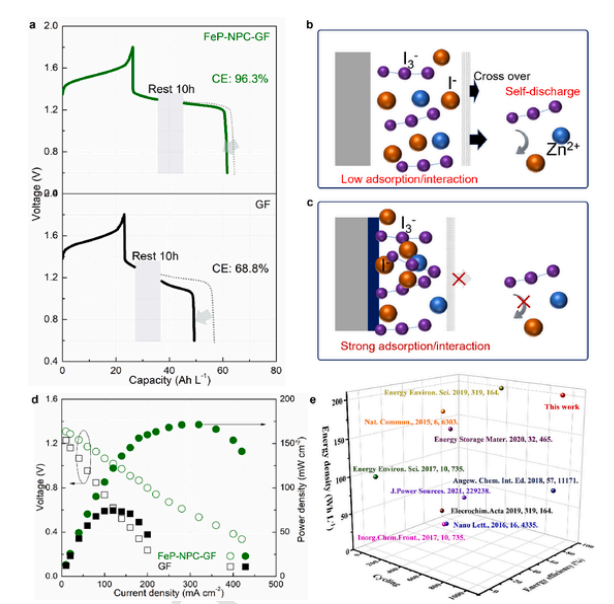
如图4所示,FeP-NPC-GF组装的Zn-I2液流电池表现出卓越的循环稳定性,这可以归因于其出色的抗自放电能力(图4a)。具体来说,经过10 h的闲置期后,带原始碳毡的充满电的 Zn-I2 液流电池仅保留了其原始放电容量的 68.8%,而且放电过电位明显增加。相比之下,当我们使用改进的FeP-NPC-GF基质时,自放电现象几乎可以忽略不计(96.3 %保留率),表明该Zn-I2 液流电池具有较高的稳定性。如图6所示,FeP-NPC-GF 的容量下降率更低(容量损失率为 7.6%,而 GF 组装电池的容量损失率为 38.7%),在 10 小时闲置期的电化学稳定性更高且电压变化极小。如图4b和c所示,阴极溶液和碳基底之间有限的界面相互作用导致(多)碘化物穿梭,碘中间体和沉积锌之间的阳极界面化学反应加速了电池容量的衰减。因此,尤其是在充电过程中,Zn(I3)2 很容易与锌发生反应,在阳极侧形成 ZnI2,这将大大降低 Zn-I2 液流电池的 CE 值。由此我们推测FeP-NPC-GF对碘/多碘化物限制和碘/多碘化物氧化还原具有双功能效应。杂原子和纳米团簇的协同作用促进了与碘/多碘化物活性物种的充分结合亲和力以及高效的液–固界面电荷转移。具体来说,杂原子可以赋予强界面化学吸附,而FeP纳米团簇则会加速电子转移,从而限制多碘化物的穿梭,增强氧化还原动力学。图 4d 显示了 Zn-I2 液流电池的放电极化和功率密度曲线。FeP-NPC-GF 使液流电池的峰值功率密度达到 172 mW cm-2,而没有原始 GF 电池的峰值功率密度仅为 74 mW cm-2。我们组装的 Zn-I2 液流电池电池的能量密度(204 Wh L-1)、循环稳定性(1000 次)和能量效率(87%)与最近发表的最先进的锌氧化还原液流电池相比毫不逊色(图 4e)。
图4。Zn-I2电池的抗自放电特性和功率密度。(a) 使用纯GF和FeP-NPC-GF对Zn-I2电池进行长时间待机测试。(b) GF和 (c) FeP-NPC-GF组装液流电池自放电容量衰减机制示意图。(d) Zn-I2液流电池的放电极化曲线。(e) 本工作的Zn-I2液流电池与其他已报道的碘基液流电池的电化学性能比较。
综上所述,我们在此报告了一种新型双功能石墨毡基质的高稳定性锌–聚碘液流电池的成功典范。在多孔N和P共掺杂碳框架内,FeP纳米团簇的良好分散促进了碘/多碘化物的高效化学吸附,并提供了电催化作用,以加快I−/I3−氧化还原,从而有效地抑制了多碘化物的穿梭,有效减轻了电池的自放电问题。因此,FeP-NPC-GF组装的Zn-I2液流电池在20 mA cm -2下的电流密度下,获得高达204 Wh L−1的理论容量,并且具有1000次循环的良好长周期稳定性,且平均CE达到创纪录的98.1%。双功能石墨毡的精心设计说明了提高锌离子流动化学性能的有效方向。Hong Zhang, Tianhang Ding, Rongqian Kuang, Ke Lu, Songtao Lu, A bifunctional electrocatalytic graphite felt for stable aqueous zincpolyiodide flow batteries, Journal of Power Sources, 2024.
https://doi.org/10.1016/j.jpowsour.2024.234798