
第一作者:石文辉&刘瀚文
通讯作者:夏宝玉&姚永刚
通讯单位:华中科技大学
成果简介
异质结构催化剂是能量转化和储存的关键。然而,在保持对不同成分的精确控制的同时,扩大它们的合成规模是一项挑战。为此,华中科技大学姚永刚和夏宝玉团队介绍了金属和碳之间的氧化电位差异作为设计多元素异质结构的热力学因素。为实现多元素异质结构催化剂的一步合成和连续生产,开发了一种卷对卷碳热冲击技术。该方法可合成多种异质结构催化剂,从单一元素到高熵合金、氧化物及其组合。此外,动力学可调性可以精确控制元素分布,并有助于识别不同的元素区域,以指导异质结构催化剂的精细定制。作为概念验证,作者演示了快速筛选PtCo@La-TiO2用于碱氢演化。本研究提出了一种快速合成、筛选和连续生产多元素异质结构催化剂的先进技术。
感谢华中科技大学姚永刚和夏宝玉团队(第一作者:石文辉)供稿!
本文所用
AEM电解槽
由武汉之升新能源有限公司提供

研究背景
异质结构多元素催化剂已广泛应用于各种工业过程中。而活性金属主要作为催化剂,调节吸附和关键的反应步骤,氧化物载体起着至关重要的作用,而不仅仅是分散和稳定。通过相互作用,如强金属-支撑相互作用,氧化物作为助催化剂,增强活性和稳定性。然而,传统的制造方法,如浸渍、吸附、沉淀或离子交换,依赖于多步骤过程,往往导致活性组分之间的界面定义不清。由于潜在的金属聚集和不均匀分布,后续的处理步骤,包括洗涤、干燥和煅烧,可能进一步损害活性位点的可达性和对金属-载体相互作用的控制。虽然在开发多元素合金和氧化物催化剂方面取得了进展,但对其异质结构中热力学相的形成和特定界面的设计仍然知之甚少。这种知识差距限制了许多有前途的催化剂的可扩展性和实际应用。越来越复杂的多元件系统和缺乏有效的大规模制造方法进一步加剧了这一限制。
核心内容
1. 异质结构催化剂的卷对卷合成
图1a简要地说明了通过卷对卷CTS方法快速和跨规模地制造多元素异质结构催化剂。合金和氧化物组成的混合前驱体滴在碳布(CC)上,对其进行诱导焦耳加热。高温CTS使金属和氧化物的合金化均匀,但金属和氧化物之间的自动相分离发生,这是由金属和碳之间的氧化电位差引导的。多元素异质结构催化剂利用原位快速高温工艺,在瞬间形成界面紧密的催化剂。
如图1b所示,装载金属盐前驱体的商用CC以7 m min-1的速度释放,通过两个石墨辊。在两个石墨辊之间的CC上施加大电流,引起焦耳加热,以提高催化剂合成温度。由于采用电加热,可以通过调节施加的电流轻松调节温度,加热持续时间受轧制速度和两个石墨辊之间距离的影响(图1c)。同时,将焦耳加热后的自支撑催化剂收集在滚轮上。如图1d所示,在10 s内连续制造出一个较大的电极(10 × 100 cm2)。与传统的多步合成异质结构催化剂相比,作者的方法不仅可以在一步(~0.5 s)内快速制备催化剂,而且得益于高温CTS优异的合金化能力和元素氧化势的指导,作者的方法可以普遍合成从单一元素到高熵合金、氧化物及其异质结构的一系列催化剂(图1e)。

图1 |异质结构催化剂的卷对卷合成. a.卷对卷法合成metal@oxide异质结构催化剂示意图。b.合成过程的照片。c.在25a、30a、35a和40a的不同施加电流下,恒定速度为7m min-1,距离为15cm时对应的温度曲线。d,通过卷对卷合成方法制造的大尺寸电极(10 cm × 100 cm)的数码照片。e、异质结构催化剂的传统合成与一步法合成的比较。f,本研究的卷对卷CTS工艺与传统大规模制备工艺的效率和产率比较。
多元素异质结构催化剂也可以通过常规CTS在极短的冲击时间(<1 s)内合成。然而,这种瞬态加热允许CTS在一个批次中只适应非常微量的材料(~1 mg或1 cm2)。因此,尽管时间和节能,扩大常规CTS大规模生产是极具挑战性的。相比之下,卷对卷CTS能够以超快的制备速率(~7 m min-1)连续和直接制备大规模电极,其中空间滚动引起的冲击实现了瞬态合成和连续制造工艺之间的有效结合,表明了真正的工业级可扩展性。与之前报道的单批和大规模制备方法(如腐蚀工程、阳极氧化和电渗)相比,作者的卷对卷CTS提供了连续制备(高达7 m min-1)和更高的生产率(~116.69 cm2 s-1)(图1f)。
2. 异质结构催化剂的快速筛选
多元素异质结构催化剂具有组分可调、界面复杂等特点,特别适用于涉及多种反应中间体的多步催化反应。在这里,作者选择一个碱性HER的简单案例来演示这个概念。在碱性质子缺乏的环境中,通过水吸附和解离产生质子是制氢的先决条件,而氧化物由于其优异的亲氧性,可以促进这一过程。此外,根据Sabatier的原理,理想的HER催化剂具有中间的氢结合能,其中Pt的吸附能可以通过与传统金属合金化来优化。正如后面讨论的,快速和通用的合成使作者能够在PtM3@M2-M1Ox空间中快速筛选碱性HER的最佳多元素异质结构催化剂,其中(1)M1Ox作为助催化剂促进水吸附和解离产生质子(Volmer步骤);(2) M2掺杂M1Ox(M2-M1Ox)可以调制Volmer阶跃;(3)掺杂M3的Pt(PtM3)有利于质子吸附和氢解吸(Tafel或Heyrovsky步骤)。
最初,作者系统地研究了不同氧化物载体对HER的影响,筛选了七种异质结构催化剂(Pt@M1Ox, M1 = Cr, Nb, Ta, Ti, Hf, Zr或La)。如图2a所示,在所有Pt@M1Ox催化剂中,Pt@TiO2在电流密度为10 mA cm−2时表现出最佳的HER活性和最低的过电位,这可能归因于TiO2载体的最佳亲氧性和电导率。为了确定氧化电位(亲氧性)对HER性能的影响,建立了过电位与相对氧化电位的相关性,表明其为火山型关系。如图2b所示,Pt@Cr2O3、Pt@Nb2O5和Pt@TaO2具有较多的正相对氧化电位,而Pt@HfO2、Pt@ZrO2和Pt@La2O3具有较多的负相对氧化电位。这些样品表现出较低的HER活性,这可能是由于*OH吸附能太强或太弱,限制了HER过程。而相对氧化电位适中的Pt@TiO2则表现出较高的HER活性,说明相对氧化电位在碱性HER性能中的关键作用。
接下来,作者对氧化物载体的相对氧化电位进行微调,以提高Pt@TiO2的碱性HER性能。将相对氧化电位为正的Cr、Mo和相对氧化电位为负的La等外来物质引入载体材料中,形成Pt@M(图2a)。具体来说,Pt@La-TiO2在La掺杂后的HER活性增强,这可能是由于适度的*OH吸附能促进了HER过程。最后,根据Sabatier原理,利用过渡金属(Fe, Co, Ni和Cu)与主催化剂Pt金属进行合金制备PtM3@M2-M1Ox。第二种金属可以用H*修饰Pt的电子结构和结合能(图2a、b),从而促进质子吸附和氢解吸。这就得到了碱性HER的最佳催化剂PtCo@La-TiO2。
进一步研究了筛选得到的PtCo@La-TiO2催化剂的微观结构和形貌。首先,大规模商用CC衬底(10 × 100 cm2)显示出开放和多孔结构(约数十微米)(图2c,d),增强水和电解质浸泡,促进质量和电子传递。为了微调HER性能,作者优化了合成温度和金属和氧化物负载,作者发现最佳温度(1000°C)改善了La-TiO2的分布,0.5% PtCo和7.5%的La-TiO2是合适的剂量,具有良好的分散和催化性能。通过电感耦合等离子体质谱实验,Pt、Co、Ti和La的实际加载质量分别为0.046、0.0014、0.36和0.12 mg cm−2,其中PtCo和La-TiO2的加载量为0.474 wt.%和7.43 wt.%。
如图2e所示,层次结构的PtCo@La-TiO2催化剂附着在碳纤维表面,PtCo纳米颗粒(亮点)均匀地分散在La掺杂的TiO2岛载体上,有利于活性位点暴露。在透射电子显微镜(TEM)图像中(图2f),尺寸为~3.8 nm的高密度PtCo纳米颗粒均匀锚定在多孔的La-TiO2上,直接与碳衬底相连,有利于电子传递、电解质浸泡和气体释放。这些结果表明,从纳米尺度到微尺度的跨尺度PtCo@La-TiO2多元素异质结构催化剂已经成功制备。高倍TEM图像显示,颗粒被困在氧化物衬底中,其d-间距分别为0.225 nm和0.354 nm,这是Pt(111)和锐钛矿TiO2(101)晶格集35、36造成的,且略大于原始TiO2的面间距,表明晶格缺陷是由La掺杂引起的(图2g)。根据能谱图(图2h),Pt和Co元素被限制在纳米颗粒区域,而Ti、La和O元素分布在整个区域,进一步证实了PtCo合金纳米颗粒分布在La-TiO2载体上形成PtCo@La-TiO2。
进一步研究了催化剂的晶体结构、电子结构和化学状态。通过CTS得到锐钛矿TiO2, La掺杂在PtCo@La-TiO2中诱导了更多的氧空位(Ov)。如图19所示,与PtCo@TiO2相比,PtCo@La-TiO2中Pt 4f的结合能向更低的结合能偏移,说明La掺杂促进了电子从La掺杂的TiO2向Pt的转移,验证了PtCo与La-TiO2载体之间的强相互作用。TiO2中的杂价掺杂物La3+容易形成氧空位。对于O 1s位于531.58 eV的峰归因于氧空位。对于Ti2p,与PtCo@La-TiO2相比,PtCo@TiO2中的Ti4+的结合能更低,这可能是由于引入了更多的氧空位。此外,根据电化学阻抗谱分析,PtCo@La-TiO2在所有催化剂中表现出最小的电荷转移电阻。因此,引入的氧空位可以调整合金催化剂的电子结构,促进电荷转移,从而提高电催化性能。
为了优化大规模制造,作者将轧制速率调整为7 m min-1,并确定1000°C和5 cm距离通过卷对卷CTS产生最佳电极。为了实现再现性和均匀性,作者从大电极的中心部分分析了四个不同的区域。这些区域显示PtCo纳米颗粒在La-TiO2岛载体上分布均匀,粒径和分布一致。X射线衍射图证实了锐钛矿TiO2的存在,但没有特有的Pt信号,并且在大规模的PtCo@La-TiO2中没有观察到Cl信号,表明前驱体完全分解。重要的是,所有四个区域都表现出一致的电化学HER性能(图2i),强调了卷对卷合成方法的稳健再现性。作者进一步研究了大电极边缘部分的均匀性。显示了层次结构PtCo@La-TiO2催化剂在上下边缘的均匀分布,与中心部分一致。这种大面积均匀性主要归因于作者方法中均匀的卷对卷加热过程。
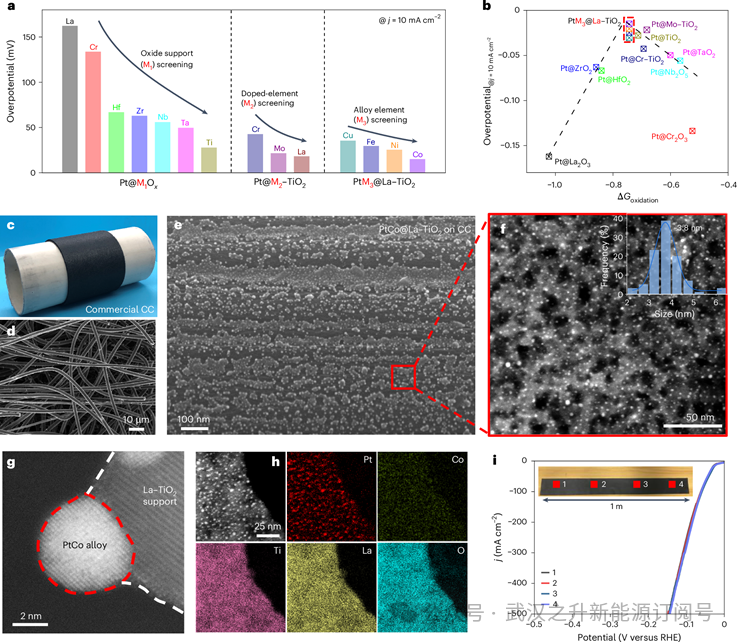
图2 | 多元素异质结构的快速筛选与结构表征. a,筛选和过电位PtM3@M2-M1Ox电流密度为10 mA cm−2。b,PtM3@M2-M1Ox过电位与相对氧化电位之间的火山图。黑色虚线表示“火山”型关系。红色虚线包含PtM3@La-TiO2中的样本,其中M3 = Cu,青色;铁、蓝色;倪,橙色;还有,丁香。c,d CC的照片. e. PtCo@La-TiO2的SEM图像。f, PtCo@La-TiO2的TEM图像。g,PtCo@La-TiO2的高分辨率TEM图像。h,PtCo@La-TiO2的高角度环形暗场扫描TEM (HAADF-STEM)和EDS图谱。i,PtCo@La-TiO2在四个不同区域的LSV曲线。
3. 异质结构PtCo@La-TiO2的催化评价
在1.0 M KOH电解液中,通过三电极结构对制备样品的电催化HER性能进行了全面评估。图3a的线性扫描伏安图(LSVs)显示,La的掺入增强了PtCo@La-TiO2的HER活性。PtCo@La-TiO2在电流密度为10 mA cm−2时表现出15 mV的商业Pt/C样活性,低于Pt/C (24 mV)、Pt (62 mV)和PtCo@TiO2 (27 mV)(图3b)。此外,PtCo@La-TiO2在−0.1 V电位下的电流密度达到189 mA cm−2,超过了商用Pt/C (80 mA cm−2)、Pt (22 mA cm−2)和PtCo@TiO2(122 mA cm−2)。值得注意的是,PtCo@La-TiO2与商业Pt/C (64.4 mV dec−1)、Pt (117.4 mV dec−1)和PtCo@TiO2(43.8 mV dec−1)相比,具有更小的Tafel斜率(19.8 mV dec−1)(图3c),表明HER动力学过程遵循高效的Volmer-Tafel机制。此外,PtCo@La-TiO2的碱性HER活性超过了最近报道的大多数其他pt基催化剂。
虽然PtCo@La-TiO2的表面积比PtCo@TiO2增加了1.3倍,但PtCo@La-TiO2的比活性仍然大于PtCo@TiO2,说明PtCo@La-TiO2优越的HER性能不仅来自于表面积的增加,还来自于内在活性的增加。此外,PtCo@La-TiO2电极的TOF值(在过电位为70 mV时为2.39 s−1)高于PtCo@TiO2 (1.34 s−1)、Pt/C (0.95 s−1)和Pt (0.26 s−1),表明PtCo@La-TiO2具有较高的内在HER活性。与具有较大电压降(212 mV)的商用Pt/C相比,PtCo@La-TiO2在100 h后表现出可忽略不计的电压降(38 mV),表明具有优异的电化学稳定性,这也可与最先进的Pt基催化剂相媲美。稳定性测试后的微观结构检查显示,与商业Pt/C中明显的颗粒聚集不同,PtCo@La-TiO2的形貌保持不变,这表明La-TiO2载体通过强金属-载体相互作用增强了PtCo纳米颗粒的稳定性。
为了阐明异质结构PtCo@La-TiO2在碱性HER中的作用,作者通过实验和理论两种方法系统地研究了水吸附、解离、质子吸附和氢脱附的催化过程。PtCo@La-TiO2电极表现出更强的疏气和超亲水特性,这要归功于其分层结构表面和亲氧La掺杂。此外,利用自旋极化密度泛函理论计算揭示了La掺杂和氧空位对PtCo@La-TiO2碱性HER增强的影响。电子从La-TiO2转移到PtCo团簇,证明了PtCo@La-TiO2中富电子PtCo团簇的形成。此外,与PtCo@TiO2相比,PtCo@La-TiO2中Pt的d带中心向费米能级偏移了0.12 eV。根据d带中心理论,d带中心的下移通常会导致H*吸附减弱,从而促进氢溢出,最终产生H2 46,47。在碱性电解质中,良好的水吸附和解离动力学是HER的先决条件,并直接决定HER的活性。氧化物载体和La掺杂的引入加强了水分子与PtCo@La-TiO2的结合。作者进一步计算了PtCo@TiO2和PtCo@La-TiO2的水解离的动能垒(图3d)。值得注意的是,PtCo@La-TiO2的水解离能垒(0.1 eV)远低于PtCo@TiO2 (0.79 eV),说明在TiO2中掺杂La原子可以促进HO-H键的断裂,形成*H中间体。PtCo@TiO2表现出较弱的吸附自由能(-0.78 eV),这是由于电子从TiO2载体转移到PtCo簇。
图3e显示了PtCo@La-TiO2通过氢溢出途径在Pt位点(青色线)和Ov位点(红线)上的HER自由能值。PtCo团簇中空位置的ΔGH*值为-0.31 eV,随后扩散到Pt原子与Ov之间的界面位置,提供ΔGH*值为0.04 eV,说明了氢的易析出。实质上,PtCo@La-TiO2表面的碱性HER涉及到Pt位点上H2O的解离、氢从Pt位点向Ov位点的溢出以及Ov位点上氢的高效解吸。因此,TiO2载体中掺杂La增强了水吸附能力,降低了水解离的能垒,促进氢从PtCo合金溢出到La-TiO2载体的Ov位点,协同增强了电催化析氢活性。
此外,为了评估PtCo@La-TiO2催化剂的应用潜力,作者组装了一个阴离子交换膜(AEM)电解槽,分别使用大规模制备的PtCo@La-TiO2(Pt质量负载为~0.05 mg cm-2)和NiFe@NF作为阴极和阳极电极(图3f)。将PtCo@La-TiO2和商用Pt/C粉末催化剂喷在碳纸上,形成S-PtCo@La-TiO2和S-Pt/C(Pt质量负载~0.3 mg cm−2)电极。所示图3 g, PtCo@La-TiO2||NiFe@NF电解槽表现出高的催化活性,在1.85 V槽压下达到1000 mA cm−2的电流密度,优于S-PtCo@La-TiO2||NiFe@NF (1.94 V@1000 mA cm−2)和S-Pt/C||NiFe@NF (2.2 V@500 mA cm−2) 电解槽。作者还组装了具有不同电极尺寸(1 × 1、2 × 2和5 × 5 cm2)的AEM电解槽,其中它们都具有相似的电解水性能,这表明作者的卷对卷制备电极在电解槽中的性能是一致的。
此外,作者还测试了PtCo@La-TiO2 ||NiFe@NF电解槽在1000 mA cm−2电流密度下的长期稳定性(图3h),该电解槽在长期运行100 h时保持了近乎恒定的槽压,没有明显的退化。然而,S-PtCo@La-TiO2||NiFe@NF和S-Pt /C||NiFe@NF电解槽的槽压在1000 mA cm−2和400 mA cm−2电流密度下分别在50 h和30 h后升高,进一步验证了PtCo@La-TiO2电极的优势。此外,采用排水法收集PtCo@La-TiO2催化剂产生的H2体积。如补充图49所示,通过比较测量和计算的产氢体积,估计法拉第效率接近90%。上述场景还使作者能够通过多元素异质结构设计(补充图50)快速优化用于恶劣碱性析氧反应(OER)的催化剂,其中FeCoNiCrMn@HfMoWZrCeOx在100 mA cm-2下的过电位最低为265 mV,并且长期稳定(~250 h无明显降解),表明多元素异质结构设计对于提高恶劣碱性OER催化性能的重要性。
图3 |PtCo@La-TiO2催化剂的HER性能。a, Pt/C、Pt、PtCo@TiO2和PtCo@La-TiO2的LSV曲线。b, Pt/C, Pt, PtCo@TiO2和PtCo@La-TiO2在10 mA cm-2的过电位和-0.1 V的电流密度。c,相应的Tafel斜率。d,不同催化剂的水解离动力学势垒。IS,初始态;TS:过渡态;FS,终态。e, PtCo@La-TiO2通过氢溢出途径在Pt原子和Ov上的HER的吉布斯自由能图。f、AEM水电解槽原理图。g和h,在1.0 M KOH溶液中,以PtCo@La-TiO2或Pt/C为阴极催化剂,NiFe@NF为阳极催化剂的AEM电解槽的极化曲线(g)和计时电位测试(h)。
4. 热力学指导与通用合成
为了阐明多元素异质结构催化剂的形成机理,作者分析了Ellingham图。在CTS过程中,元素氧化电位的显著差异促进了金属(一次催化剂)和氧化物载体(分散、稳定和辅助催化剂)之间的相分离。作者从Ellingham图中提取了每种金属元素和碳在特定温度(Temp)下的氧化电位值。然后,作者计算了金属元素与碳的氧化电位。作者确定了它作为一个热力学描述符来指导异质结构催化剂的合成。例如,如图4a所示,作者将每种元素与碳在1000°C时的氧化电位进行了比较,其中正值(用红色标记)表示易于被CTS还原和合金化,相反,更负的(用蓝色标记)表示易于氧化形成氧化物支撑。在多元素体系中,符号差异诱导金属和氧化物载体之间的自然相分离,使多元素异质结构的一步原位合成成为可能。
因此,一系列多元素合金/氧化物催化剂(如Pt@HEO-5, HEA-5@TiO2和HEA-5@HEO-5)通常在一个步骤中合成,其中原位形成的异质结构与文献中经常报道的金属-载体结构相比,具有强而紧密的界面。利用高温CTS工艺,在瞬间形成具有金属支撑界面的多元素异质结构催化剂。例如,(1)对于多元素alloy@oxide异质结构,如图4b所示,Pt、Ir、Ru、Rh和Pd信号被限制在颗粒区域内,而Ti和O信号分布在整个颗粒区域,说明PtIrRuRhPd多元素高熵合金纳米粒子(HEAs)被负载在TiO2载体上形成HEA-5@TiO2;(2)对于metal@multielement氧化物异质结构,从图4c可以看出,Zr、Ti、Nb、Hf、V和O信号分布在整个区域,Pt信号位于颗粒区域,说明Pt颗粒锚定在ZrTiNbHfVOx多元素高熵氧化物载体(HEO)上形成Pt@HEO-5;(3)对于多元素alloy@multielement氧化物异质结构,如图4d所示,EDS图谱显示PtIrRuRhPd纳米颗粒分布在ZrTiNbHfVOx载体上,形成HEA-5@HEO-5,表明多种异质结构催化剂的合成具有广泛可调的成分和金属-载体界面。
作者还观察到,增加氧分压会降低氧化电位(图4e)。这可能导致某些元素的氧化电位差在较高的氧偏压下从正变为负,从而诱导从还原态到氧化态的转变。为了验证这一效应,作者在不同的气氛下合成了PtCoWCrTi催化剂。在Ar气氛下(图4f), Pt和Co形成金属颗粒,Ti和O形成氧化物载体。值得注意的是,W和Cr同时存在于金属相和氧化物相中,导致PtCoWCr在PtCoWCr@WCr-TiOx结构中锚定在wcr掺杂的TiOx上。相比之下,在空气气氛下(图4g),Pt和Co形成金属颗粒,而W、Cr、Ti和O由于W和Cr的完全氧化形成氧化物载体,导致PtCo锚定在WCrTiOx载体上的PtCo@WCrTiOx结构。这些结果表明,氧分压对多元素异质结构的形成及其金属与氧化相间元素分布的可调性有较大影响。
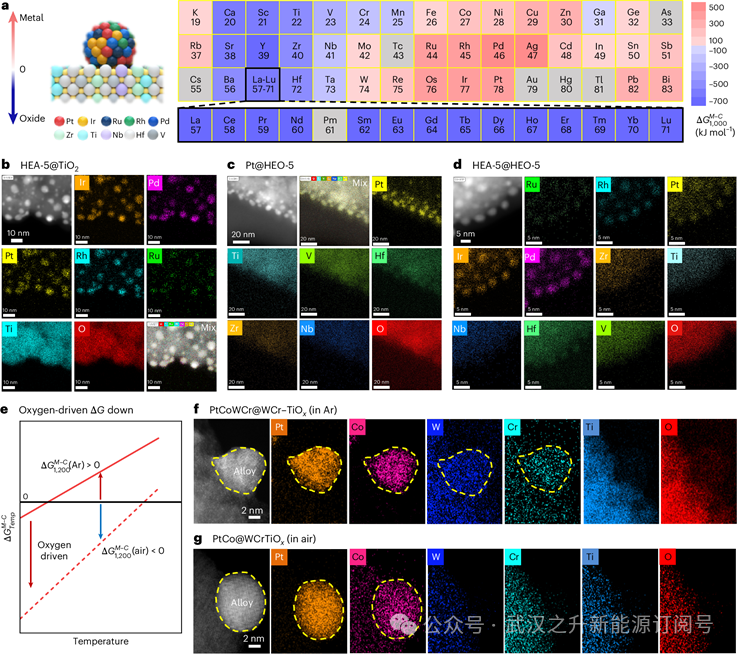
图4 | 多元素异质结构的热力学准则及一般合成. a,元素周期表中1000℃时的热力学描述符。数据来源:文献54。b-d, HEA-5@TiO2 (b), Pt@HEO-5 (c)和HEA-5@HEO-5 (d)的HAADF-STEM和EDS测绘图像。e, Ar和空气中计算的变化。f,g, PtCoWCr@WCr-TiOx (f, in Ar)和PtCo@WCrTiOx (g, in air)的HAADF-STEM和EDS图谱。
5. 动态可调性和元素指导图
虽然金属和氧化物载体之间的界面相互作用和重建已经被广泛研究了,但异质结构催化剂中元素分布的调控,特别是在多元素体系中,仍然相对未被探索。本文揭示了的温度敏感性,为通过动力学操作精细控制多元素异质结构中的元素分布提供了坚实的基础。作者发现,金属/氧化物的行为可以很大程度上受到CTS温度的影响,特别是对于值接近零的元素,允许它们经历具有动态可调性的还原或氧化状态。元素分布可以通过精确的CTS工艺动态调整,以实现动态可调元素(DTE),其中这些元素可以随着CTS温度的升高从氧化物迁移到合金区域(图5a-c)。
如图5d-f所示,在不同温度下,Pt和Pd被限制在金属颗粒区域的特定区域,而Ti和Zr信号则分布在整个氧化物载体中。有趣的是,Fe和Ni信号与温度有关:(1)在高温CTS(1100°C)期间,它们主要位于颗粒区域;(2)在低温CTS(900℃)过程中,它们分布在氧化物载体中;(3)在中温CTS(1000℃)过程中,它们不仅分散在颗粒区,而且也位于氧化物载体中,表明它们在金属纳米颗粒和氧化物载体之间具有动态可调性,从而合成了PtPd@FeNiTiZrOx、PtPdFeNi@TiZrOx和PtPdFeNi@FeNi-TiZrOx。此外,利用X射线光电子能谱分析了金属态和氧化物态Ni和Fe的含量。如图5g所示,金属Ni和Fe含量随温度升高而增加,与EDS作图结果一致。
此外,Fe, Co, Ni, Mo, W和Sn位于碳氧化曲线附近,这表明它们的金属/氧化物行为可以受到CTS温度的影响,使它们能够进行动态可调性的还原或氧化。例如,设计了一种更灵活、可调节的多元素alloy@oxide异质结构催化剂。如图5h所示,Pt, Ir, Ru, Rh和Pd元素被限制在金属颗粒区域的特定区域,而Zr, Ti, Nb, Hf, V, Ce和O信号分布在整个氧化物支撑体中。有趣的是,Fe, Co, Ni和Mn信号主要位于颗粒区域,剩余的一些信号分布在整个区域,表明它们在金属纳米颗粒和氧化物载体中都具有动态可调性,导致PtIrRuRhPdFeCoNiMn锚定在feconimn掺杂的ZrTiNbHfVCeOx上(HEA-9@HEO-10)。这些异质结构材料具有广泛的组分和多个活性位点,非常适合于各种多步催化反应。基于上述结果,作者将元素分为三类:(1)金属/合金区(在任何温度下),(2)氧化物区(在任何温度下)和(3)DTE区(随着温度的升高从正变为负)(图5i),其中开发了精细定制多元素金属-氧化物异质结构催化剂的元素引导图。

图5 |多元异质结构的动态可调性与单元导向性图. a CTS工艺的动态可调温度(红色,~ 1100℃;藏蓝色,~ 1000℃;黑色,~900°C)。b、的温度依赖关系。c、元件动态调节示意图。d-f, PtPd@FeNiTiZrOx (d), PtPdNiFe@FeNi-TiZrOx (e)和PtPdNiFe@TiZrOx (f)的HAADF-STEM和EDS图谱。g,不同温度(900,1000和1100℃)下Ni0和Fe0的价态含量。h, HEA-9@HEO-10的HAADF-STEM和EDS制图图像。i、多元素异质结构催化剂元素导向性图:(1)金属/合金区,(2)氧化物区,(3)DTE区。
结论展望
综上所述,作者揭示了多元素异质结构的形成机理,并提出了金属和碳之间氧化电位的差异,作为指导多元素异质结构设计和合成的热力学描述符。为实现多元素异质结构催化剂在~7 m min-1连续快速大规模制备,开发了卷对卷CTS技术。在此指导下,合成了一系列多元素异质结构催化剂,包括HEA-5@TiO2、Pt@HEO-5、HEA-5@HEO-5和HEA-9@HEO-10。此外,对元素分布进行动力学调控,进一步确定不同元素区(金属区、氧化物区或可调区),合理构建多元素异质结构。本研究探讨了多元素异质结构催化剂的快速合成和跨规模制造的热力学准则和先进制造方法,在各种多步骤催化过程中呈现出有希望的可扩展性和适用性。
文献信息
Wenhui Shi, Hanwen Liu, Jianwei Zhang, Shenyu Shen, Yuhan Wang, Yaqing Guo, Kaihang Yue, Zihui Liang, Hao Zhang, Lei Zhang, Fatang Tan, Zhiqiang Liang, Yingjun Liu, Yaqiong Su, Dong Su, Yunhui Huang, Bao Yu Xia & Yonggang Yao
Roll-to-roll synthesis of multielement heterostructured catalysts, Nature Synthesis. (2025)
DOI: https://doi.org/10.1038/s44160-025-00758-y