
第一作者:张敏
通讯作者:付乾
通讯单位:重庆大学

气体扩散电极(GDE)作为膜电极组件(MEA)反应器的关键组成部分,凭借其低欧姆电阻和优异传质性能,在大规模电催化二氧化碳还原(CO₂RR)中展现出广阔前景。然而,传统碱性或中性电解质体系中由OH⁻引起的碳酸盐化及析氢等问题严重制约了其工业化进程。采用酸性介质策略可有效缓解碳酸盐形成,但强质子环境也易导致析氢反应(HER)占主导,显著降低CO₂还原效率。本文,重庆大学付乾教授团队研究了在纯酸性条件下的MEA型CO₂RR性能提升策略。该研究通过配位相互作用与亲核取代反应,在催化剂表面构建了带有R₄N⁺官能团的交联聚电解质涂层。该涂层不仅提供正电荷以增强局部电场、稳定关键中间体,还显著提高了CO₂RR催化活性和选择性。进一步借助原位衰减全反射–表面增强红外吸收光谱(ATR-SEIRAS)分析,研究揭示了聚电解质修饰对界面氢键网络的重构与增强机制,突出了聚电解质工程在优化酸性CO₂RR界面微环境中的重要作用。
相关成果以“Surface-immobilized cross-linking tetraalkylammonium cations networks mitigate hydrogen evolution for pure acidic CO2 reduction in proton-exchange membrane electrolyzers”为题发表在Journal of Energy Chemistry上期刊上。
感谢重庆大学付乾教授团队
(第一作者:张敏)供稿!
本文所用
膜电极(MEA)型CO₂RR反应器
由武汉之升新能源有限公司提供


基于气体扩散电极(GDE)的膜电极组件(MEA)反应器具有低欧姆电阻和优异传质性能,被广泛认为是实现大规模二氧化碳还原(CO₂RR)的理想电解槽构型。早期研究多采用阴离子交换膜(AEM),其碱性环境有利于高选择性和高法拉第效率(FE);然而,碳酸氢盐的形成与迁移会导致严重的CO₂穿透问题,显著限制单程转化率(SPC)(图1A)。为解决该问题,研究者开发了基于质子交换膜(PEM)的CO₂RR系统。在该体系的酸性阴极环境中,生成的碳酸盐可被质子(H⁺)迅速再生为CO₂,从而有效防止CO₂向阳极穿透(图1B)。近年来,酸性CO₂RR研究普遍引入碱金属阳离子以调控阴极界面微环境。这些阳离子富集于亥姆霍兹外平面(OHP),既可增强局部电场、稳定反应中间体,促进CO₂RR,又能屏蔽阴极电场、抑制H⁺迁移,缓解析氢反应(HER)竞争。但在高电流密度下,阳离子持续积累一旦超出H⁺缓冲容量,便会引起盐沉积,造成GDE孔道和阴极微通道堵塞,最终导致电解槽运行失败。近期提出的电催化剂表面有机阳离子修饰策略,可在避免碳酸盐沉积的前提下实现类似的界面调控:提供正电荷以增强局部电场、稳定关键中间体,提高CO₂RR性能(图1C)。在与纯酸/水阳极配对时,H₃O⁺可经PEM持续迁移至阴极。由于CO₂还原动力学缓慢,界面处H₂O和H⁺均可能被还原发生HER。因此,维持界面氢键网络完整性以减少游离水还原,同时增加OHP区正电荷以屏蔽电场、阻碍H⁺向表面迁移,将有助于在纯酸性PEM体系中促进CO₂还原反应。
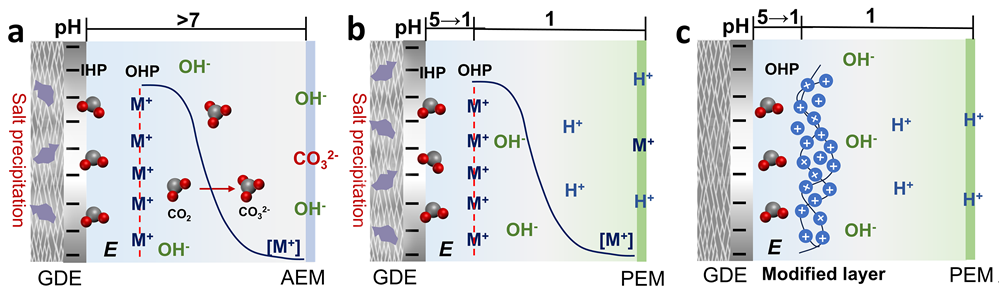
图1.不同反应介质和构型CO₂RR示意图:(a) 中性或碱性阴离子交换膜 (AEM)的MEA;(b) 有碱金属阳离子的酸性质子交换膜(PEM)的MEA;(c) 催化剂表面固定有机阳离子的纯酸性PEM的MEA。
在此,重庆大学付乾教授团队通过在催化剂表面利用配位相互作用与亲核取代反应,成功固载了带有R₄N⁺官能团的聚电解质,实现了纯酸性膜电极组装(MEA)系统中高效的CO₂电解。该团队采用聚二甲基二烯丙基氯化铵(PDDA)对银电极进行包覆,得益于PDDA中高密度季铵基团所带来的强正电场特性,所制得的Ag@PDDA电极在以H₂SO₄为阳极电解液的MEA电解槽中显著抑制了析氢反应(HER),并有效促进CO₂向CO的转化。在100 mA·cm⁻²的电流密度下,该电极对CO的法拉第效率(FE)高达86%,单程转化率(SPC)达到72%,其性能远超以往基于碱金属阳离子体系的报道。通过对比不同界面的原位衰减全反射–表面增强红外吸收光谱(ATR-SEIRAS)发现,Ag/QAPPT与Ag@PDDA界面处的孤立水(i-H₂O)占比均低于Ag及Ag/CTAB界面,表明聚电解质修饰增强了氢键网络连通性。更重要的是,Ag@PDDA因具备更高的电荷密度,可建立更强界面电场,不仅有效阻抑H⁺迁移,还促进CO₂加氢生成关键中间体COOH,从而高效推动CO形成(图2)。该研究突显了聚电解质工程在纯酸性CO₂还原反应界面调控中的重要作用,为设计高效酸性CO₂电解系统提供了新思路。
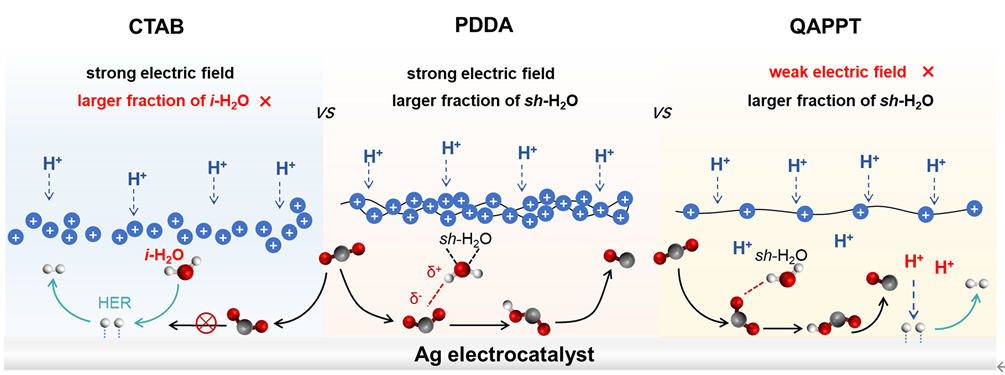
图2. PDDA结构不仅能抑制 H⁺的迁移并阻隔H₂O与催化剂的直接接触,还能在纯酸性体系中促进从CO₂到关键中间体 *COOH的生成。
1.PDDA层制备与表征
为了确保PDDA有机阳离子层在电解过程中稳定附着于催化剂表面而不被冲刷流失,作者设计了一种交联网状结构,通过配位与亲核取代过程成功制备了PDDA包覆的银电极(Ag@PDDA)。扫描电子显微镜(SEM)图像表明,原始银纳米颗粒表面光滑,粒径约为60 nm;而Ag@PDDA则呈现出典型的多孔有机聚合物网络形貌。透射电子显微镜(TEM)进一步显示,颗粒表面覆盖有一层平均厚度约2.0 nm的均匀聚合物包覆层。X射线光电子能谱(XPS)和傅里叶变换红外光谱(FTIR)分析均检测到与PDDA聚电解质结构一致的特征峰,证实其成功复合。Zeta电位测试结果表明,材料表面带正电,且Ag@PDDA的电势明显高于纯共聚物,这主要归因于交联过程中季铵氮(N⁺)官能团的生成所带来的更高正电荷密度。
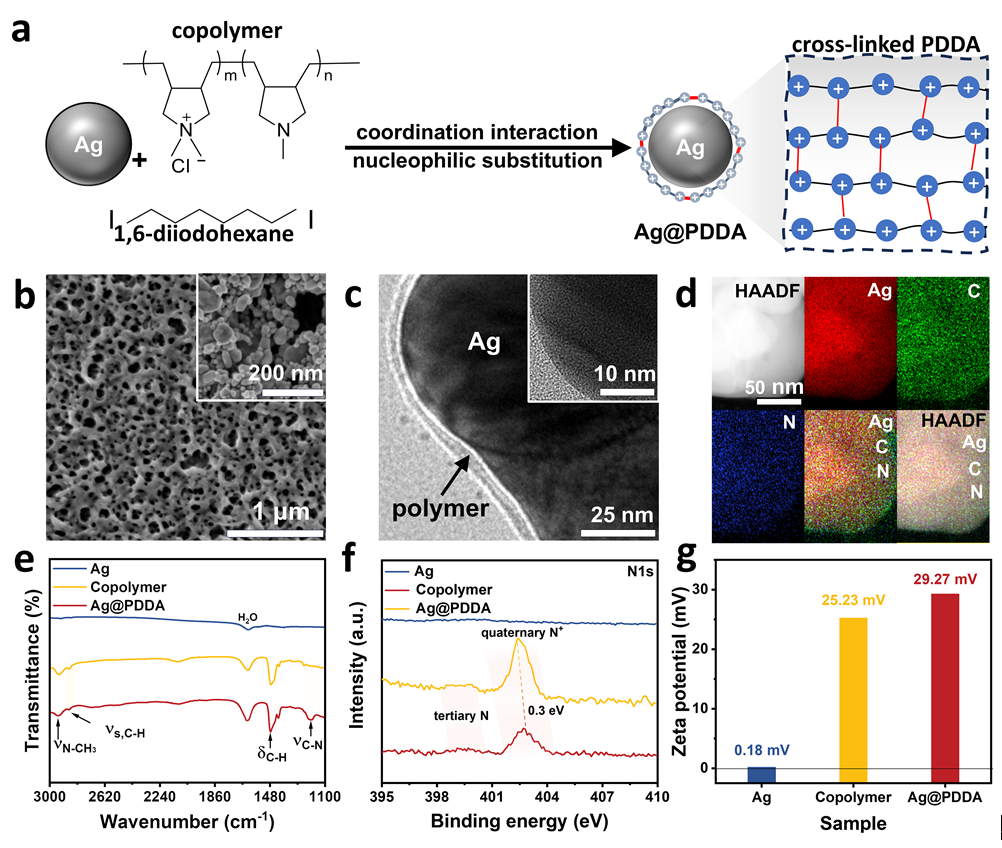
图3. (a)PDDA聚电解质在Ag上交联;(b,c)Ag@PDDA的SEM和TEM图像,插图是纯Ag颗粒;(d)HAADF-STEM和相应的EDS图谱Ag@PDDA;(e,g)银、共聚物和Ag@PDDA的FTIR光谱、N1s图谱和zeta电位。
2.酸性CO2RR性能
在50–300 mA/cm²的宽电流密度范围内,Ag@PDDA电极的CO法拉第效率(FE)显著高于未修饰的Ag电极及物理混合的Ag/PDDA样品。未修饰的Ag电极几乎未产生任何CO₂还原反应(CO₂RR)产物,而Ag/PDDA的CO法拉第效率仅约为20%。相比之下,Ag@PDDA在200 mA/cm²电流密度下实现了高达86%的CO法拉第效率。该膜电极组件(MEA)在200 mA/cm²和300 mA/cm²电流密度下的全电池电压分别为3.37 V和4.17 V,均低于以往文献报道的数值。系统在100 mA/cm²下连续运行28小时的稳定性测试表明,电池电压与高CO法拉第效率均得到良好维持。本研究在纯酸性MEA中实现的CO₂RR电流密度与CO法拉第效率均优于以往报道结果,甚至超过含碱金属阳离子的体系,突显了有机阳离子修饰策略在促进CO₂还原反应中的重要作用。
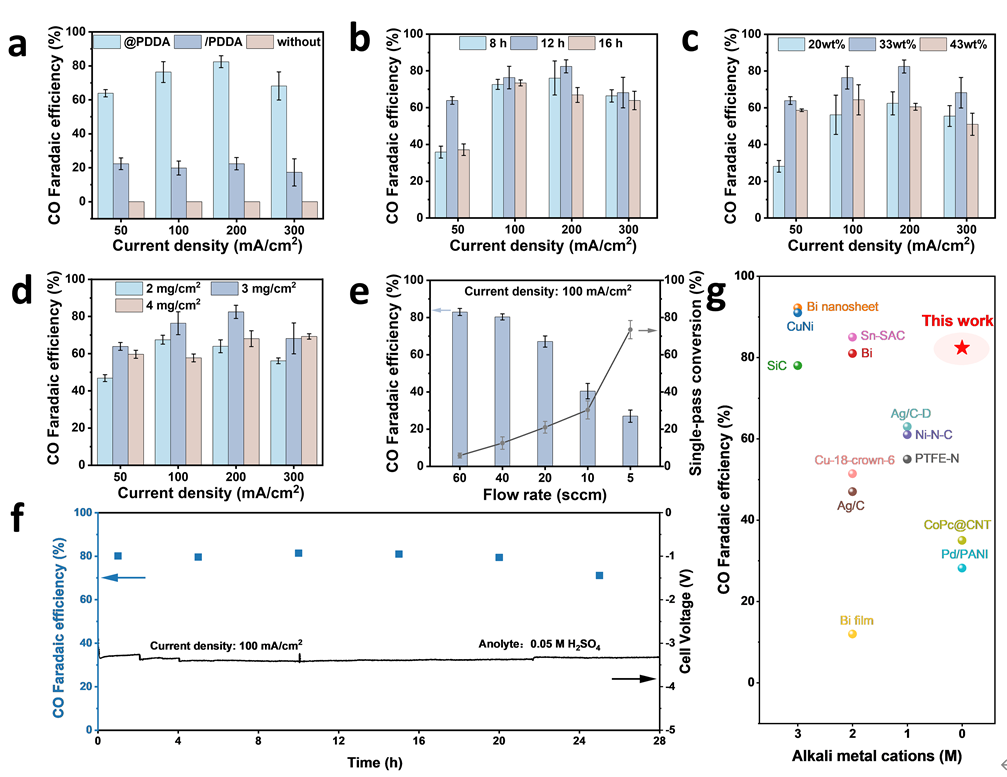
图4. H2SO4供给MEA的CO2RR:(a)Ag@PDDA、Ag/PDDA和纯Ag催化剂的FECO;(b)PDDA交联时间对FE CO的影响;(c)PDDA层负载量对FE CO的影响;(d)催化剂层负载量对FE CO的影响;(e)Ag@PDDA的FE CO和SPC;(f)Ag@PDDA运行稳定性;(g)有无碱金属阳离子的酸体系FEC1比较。
3.酸性CO2RR界面过程与机理
为探究有机阳离子聚合物在纯酸性体系中促进CO₂还原反应(CO₂RR)及抑制析氢反应(HER)的作用机制,本研究选取了两种典型电解质——聚合物电解质季铵化聚(N-甲基哌啶-co-三联苯)(QAPPT)和单分子电解质十六烷基三甲基溴化铵(CTAB)对银催化剂进行改性,并在0.05 M H₂SO₄进料的MEA体系中进行性能评估。如图5A所示,Ag/QAPPT与Ag/CTAB的CO法拉第效率(FE₍CO₎)均显著低于Ag@PDDA,且随电流密度上升迅速下降,表明仅Ag@PDDA能有效抑制HER并促进CO生成。由于QAPPT的阳离子密度远低于PDDA,其调控电场能力有限,可能导致更多H⁺迁移至银表面(图5B)。而CTAB作为表面活性剂,其疏水烷基链与亲水季铵头基在偏压下易发生迁移并富集于催化剂界面,破坏界面水分子氢键网络。
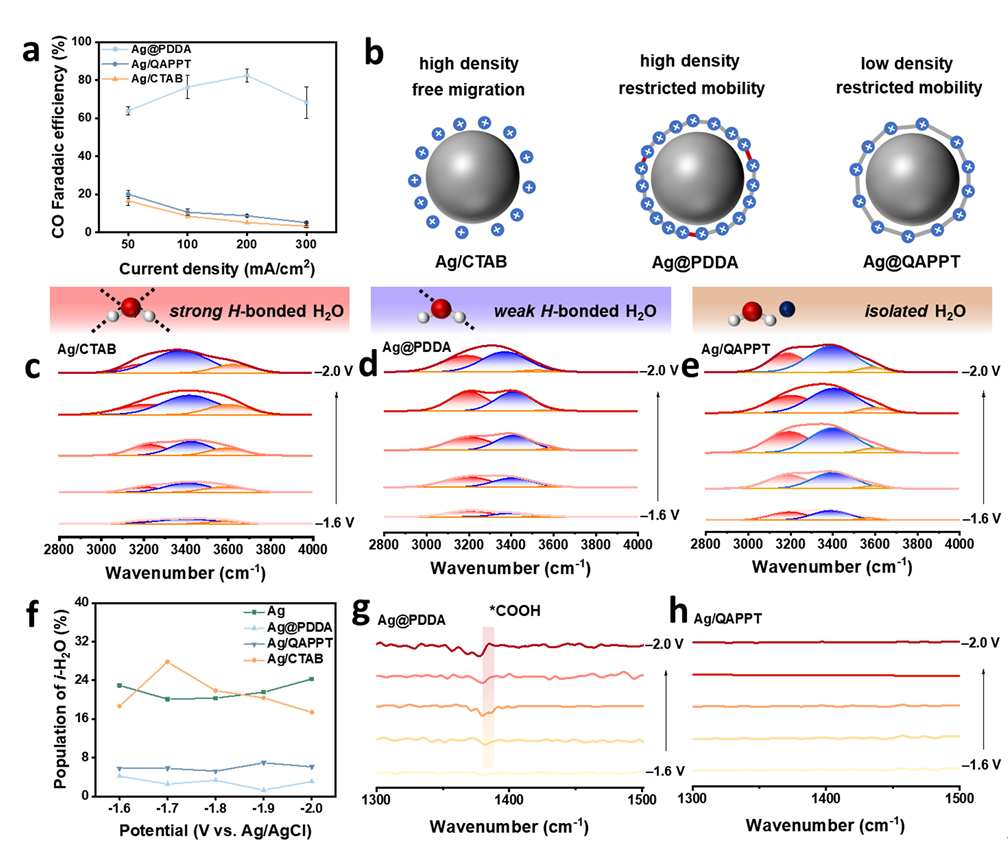
图5.Ag/有机阳离子在H2SO4进料MEA中的CO2RR:(a)Ag@PDDA、Ag/QAPPT和Ag/CTAB电极的FECO;(b)PDDA、QAPPT和CTAB在Ag表面的分布示意图;不同电位下CO2饱和0.05 M H2SO4溶液中的原位FTIR光谱:(c-f)2800-4000 cm-1不同类型的界面水;(g,h)在1300-1500 cm-1处的关键中间体。
通过原位红外光谱分析发现,与Ag/CTAB相比,Ag@PDDA和Ag/QAPPT界面处的ν(O–H)振动谱带明显向低波数位移。经高斯分峰拟合,该吸收带可分解为3600 cm⁻¹、3450 cm⁻¹和3200 cm⁻¹三个峰,分别对应孤立水(i-H₂O)、弱氢键水(wh-H₂O)和强氢键水(sh-H₂O)(图3C–F)。在Ag/CTAB界面,CTAB的迁移性导致氢键网络紊乱,i-H₂O比例较高,此类水分子更易解离产生吸附氢,引发严重HER。相反,Ag@PDDA与Ag/QAPPT保持了氢键网络的连续性,sh-H₂O占主导,水分子解离能力较低,从而抑制HER。
尽管CTAB破坏氢键网络阻碍了H⁺的传输与还原,但其引起的高比例i-H₂O仍导致水分子解离并加剧HER。对于Ag/QAPPT,大量H⁺经氢键网络传输至聚电解质界面并接触催化剂,但由于其阳离子密度较低、所产生电场较弱,无法有效屏蔽H⁺,因而仍发生明显析氢。而PDDA凭借高阳离子密度形成强电场,既可抑制H⁺向催化剂迁移,又能稳定关键反应中间体。这也解释了为何在Ag@PDDA电极表面仅检测到微弱的*COOH信号——表明在该体系中同时抑制H₂O与H⁺的还原是实现高效CO₂RR的关键。
4.纯水电解应用
为降低设备腐蚀并提高规模化应用潜力,本研究采用纯水作为电解质进行CO₂还原反应(CO₂RR),并系统评估了其在不同温度下的性能。在20°C条件下,体系在50 mA/cm²和300 mA/cm²电流密度下分别实现了35%和30%的CO法拉第效率(FE),但需承受较高的工作电压,这主要归因于传质限制与较差的电荷传输性能。为改善反应动力学,将操作温度升高至60°C,以提高离子迁移率并降低欧姆阻抗。该策略显著见效:电解槽在300 mA/cm²电流密度下的槽电压由20°C时的6.5 V降至4.4 V。
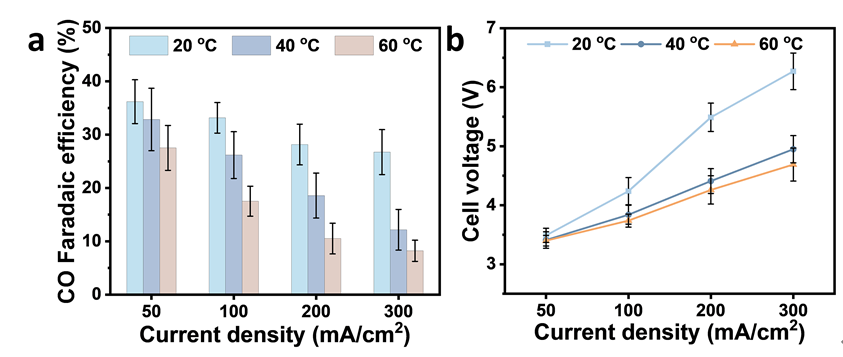
图6.不同温度下纯水环境Ag@PDDA的电催化CO2RR性能:(a)CO FE;(b)电池电压。
本研究首次采用骨架固定阳离子基团的聚电解质对银电极进行表面修饰,系统探究了接枝R₄N⁺有机阳离子在纯酸性条件下对CO₂还原反应(CO₂RR)的显著促进作用。结果表明,Ag@PDDA电极表现出优异的电催化性能:在100 mA·cm⁻²电流密度下,CO法拉第效率高达86%,单程转化率达72%,并可在28小时内保持稳定运行,充分证明了PDDA修饰层在纯酸性MEA体系中对CO₂RR的高效促进效果。通过结合电化学测试与原位ATR-SEIRAS表征,对不同R₄N⁺基电解质修饰电极的综合分析表明,PDDA修饰层凭借其富正电荷的外层和富氮杂化的内层结构,不仅有效抑制H⁺迁移、阻隔H₂O分子与Ag催化剂表面的直接接触,还在纯酸性环境中协同催化CO₂向CO转化。与Ag/QAPPT和Ag/CTAB相比,Ag@PDDA因其更强的界面电场和更低的孤立水比例,在纯酸性乃至纯水MEA体系中均展现出卓越的CO生成能力。该研究不仅凸显了电解质界面结构在调控电催化反应中的关键作用,也为质子交换膜电解技术在实际CO₂还原应用中的发展提供了重要推动。
Min Zhang, Zengyi Tan, Mufan Xing, Yang Wang, Xun Zhu, and Qian Fu. Surface-immobilized cross-linking tetraalkylammonium cations networks mitigate hydrogen evolution for pure acidic CO2 reduction in proton-exchange membrane electrolyzers. Journal of Energy Chemistry (2025). https://doi.org/10.1016/j.jechem.2025.08.050