
第一作者:刘旭
通讯作者:何刚
通讯单位:西安交通大学
成果简介
本研究首次提出了一种基于手性调节和氢键介导的溶剂化工程策略,利用邻位二羟基导向的氢键网络重构溶剂化结构,成功合成了具有差异化水溶性的手性紫精阳极电解质(R/S–异构体:2.75 ± 0.01 M;RS–消旋体:1.66 M)。这种氢键诱导的溶剂化“铠甲”赋予了电解质pH自适应的亲核屏蔽能力和碱性耐受性(pH < 11),从根本上抑制了氢氧根离子介导的降解路径。对称电池实现了卓越的循环耐久性——在3652个循环(57天)中保持了99.42%的容量保持率。此外,在1 M R/DMAP-TEMPO基水系有机液流电池(AORFBs)中验证时,该电解质在超过500个循环中实现了100%的容量保持率,显著优于阳离子[(NPr)2V]Cl4(94.92%)和阴离子(SPr)2V(65.49%)类似物。本工作通过手性调节的氢键介导溶剂化工程策略,验证了Kg-Ah级电池技术,为中性AORFBs从分子设计到工程化转型提供了完整的理论基础和技术指导。
相关成果以“Machine learning-guided solvation engineering of chiral viologens for durable neutral aqueous organic flow batteries”为题发表在Angewandte Chemie International Editione期刊上。
感谢西安交通大学刘旭博士(第一作者)校稿!
本文所用
5片分体式液流电池电堆
由武汉之升新能源有限公司提供

研究背景
人工智能(AI)技术在材料科学领域已展现出颠覆性潜力,并已成功应用于药物设计、催化材料和金属合金等领域,其预测准确性和研发效率远超传统的试错方法。基于拓扑图结构的固有特性,分子结构与性质之间的关系可通过图神经网络(GNNs)进行数学建模,从而实现从分子描述符到目标性质的预测。尽管人工智能赋能的材料研发范式已在多个领域取得突破性进展,但针对AORFB电解质设计的系统性研究仍显不足,且兼具溶解性与抗降解稳定性的多目标协同优化框架尚未建立。可预见的是,机器学习驱动的高性能紫精系设计将实现溶剂化结构的深度优化,从而同步提升溶解性和稳定性特征。
基于上述考量,本研究整合了1300余份关于AORFBs的历史文献数据集,建立了中性AORFBs的溶解度-稳定性协同优化数据库。通过充分利用大型语言模型(LLMs)在材料化学领域的丰富先验知识,结合文本嵌入技术对文献信息进行深度挖掘与分析,从而提升预测可靠性。此外,作者团队提出了一种手性导向的氢键介导溶剂化工程策略,通过立体专一性邻位二羟基配位重建氢键网络,以优化溶解度与稳定性。结合多尺度表征技术(溶剂化自由能计算、晶格能模拟、分子动力学及拉曼光谱),本研究阐明了纯对映体与外消旋体溶解行为的结构-性质关系,并揭示了pH依赖性形成的氢键“溶剂化装甲”可抵抗碱性降解。采用手性紫精负极电解液与N,N-二甲基吡啶改性TEMPO(DMAP-TEMPO)正极电解液构建的AORFBs实现了0.1~2.5 M梯度下的容量-浓度解耦;对称电池在3652次循环中保持结构完整性,未出现容量衰减。公斤级紫精合成及电堆级验证标志着向工业应用的关键过渡,为推进AORFBs从实验室原型到电网规模应用提供了理论和技术基础。
核心内容
1.利用大型语言模型进行分子结构分析与预测
 图1.机器学习指导手性紫精的分子设计。a)由大型语言模型驱动的文献信息提取流程;b)跨模型合成的应用领域、官能团及可行性评级分布;c)中性AORFBs手性紫精的分子工程化改造。
图1.机器学习指导手性紫精的分子设计。a)由大型语言模型驱动的文献信息提取流程;b)跨模型合成的应用领域、官能团及可行性评级分布;c)中性AORFBs手性紫精的分子工程化改造。
通过使用关键词“Viologen”和“AORFB”,模型系统性地识别了1300篇高度相关的出版物,构建了一个关于中性水性AORFBs的专用数据库。利用大型语言模型(LLMs),具体包括Qwenmax-latest、Moonshot-V1-32 K和GLM-4-Long APIs,通过结构化提示执行了自动化文献分析(图1a)。将“可用性报告”的文本内容通过Qwen3-Embedding模型向量化为1024维嵌入向量。在领域特定指导的引导下,模型利用材料科学知识库,将羟基官能化的手性紫精衍生物确定为最优候选物,展现出更优异的电化学和物理化学特性(图1b)。
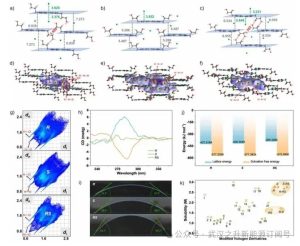
图2.手性紫精的X射线单晶结构,包括a)R型、b) S型和c)RS型;d) R型、e)S型和f) RS型的Hirschfeld表面,显示O─H···Cl和O─H···O键;g)手性紫精的二维指纹图;h)对称手性紫精衍生物的圆二色谱;i)手性紫精与水的接触角;j)手性紫精的溶剂化自由能和晶格能;k)不同取代基构型改性的紫精衍生物的溶解度比较分析。几何符号和颜色区分改性类型:紫色六边形(邻位取代)、蓝色圆圈(间位取代)、绿色方块(对位取代)、棕色三角形(末端N-烷基化)和橙色菱形(手性紫精)。
2. 合成与结构表征
基于人工智能预测的手性紫精结构,本研究设计了135种以上对称与不对称候选物,利用溶剂化效应抵抗OH−攻击,从而防止烷基链解离(图1c)。二羟基烷基衍生物因其天然前体的丰度,在所有立体化学构型中均展现出更优的商业可行性。作为电中性的氢键供体/受体,羟基通过与水建立双向氢键网络,通过增强溶质-溶剂相互作用来提高电解质溶解度。此外,本研究构建了新型手性紫精结构:通过一锅水热法将3-氯-1,2-丙二醇及其互补手性对映体功能化于4,4-联吡啶核心骨架,产率达85%–92%。通过20 L夹套反应器的连续流合成验证了可扩展性,实现了千克级生产(2.5 kg)。结构验证采用了协同分析技术:单晶X射线衍射(SC-XRD)、1H/13C核磁共振(NMR)光谱和高分辨率质谱(HRMS),共同确认了手性紫精的化学完整性和立体化学精度。此外,圆二色光谱分析显示,手性紫精对异构体和外消旋体的圆偏振光具有明显的吸收差异(图2h)。Hirschfeld表面分析显示分子体系内存在明显的Cl···H-O和O···H-O氢键相互作用(图2d-f)。此外,外消旋的二维指纹图谱显示出与C-H···π相互作用对应的明显尖峰,表明外消旋体具有更高的分子堆积效率(图2g)。此发现进一步证实,外消旋晶体结构相较于其异构体具有更强的分子间相互作用,从而导致更高的晶格能。
3. 理化性质
手性紫精的理化特性表征(包括溶解度、电导率和动态粘度)是指导电池性能优化的关键指标。R–异构体(2.75 M)和S-异构体(2.76 M)紫精的溶解度明显优于RS外消旋体(1.66 M),对应着体积容量(约74 Ah L−1)和能量密度(90 Wh L−1)的提升。不对称手性紫精表现出类似的溶解度趋势,表明立体构型是溶剂化效率的主要决定因素。接触角测量(R:37.2°,S:38.5°)的表面润湿性(图2i)分析显示,与外消旋体(RS:55.7°)相比,接触角降低了约30%。热力学分析确定了晶格能(R:−427.52 kJ mol−1,S:−420.30 kJ mol−1,RS:−395.33 kJ mol−1)和溶剂化自由能(R:−26.78 kJ mol−1,S:−26.90 kJ mol−1,RS:3.66 kJ mol−1)协同调控溶解行为差异(图2j)。降低的晶格能意味着溶质间内聚相互作用减弱,从而促进晶格失效。相反,更大的绝对溶剂化自由能反映了热力学上更倾向于的溶剂-溶质配位。对改性紫精(N-烷基化及邻/间/对位取代类似物)的比较评估证实,手性诱导的溶剂化增强优于传统功能化策略(图2k)。
通过循环伏安法(CV)、微分脉冲伏安法(DPV)和线性扫描伏安法(LSV)技术,作者团队研究了手性紫精的电化学行为。所有化合物均表现出两个明确且高度可逆的氧化还原电对,其峰间距(Ep)约为0.39±0.02 V,对应E1/2电位为-0.37 V和-0.76 ± 0.01 V(图3a)。与DMAP-TEMPO(0.86 V vs. NHE)作为正极电解液配对,组装的电池可实现约1.23 V(单电子转移)和1.62 V(双电子转移)的工作电压(图3b)。
4. 耐碱性分析
先前研究表明,末端N-烷基化的紫精类化合物在碱性条件下易受双分子亲核取代反应的亲核攻击,主要生成单向脱烷基化衍生物。然而,pH波动如何调节其固有分子稳定性在机制上仍未阐明。因此,系统评估碱性胁迫下的结构稳定性,为pH耐受性电解质的分子设计原则提供了重要见解。浓度依赖性pH比较分析显示三种紫精衍生物具有不同的质子交换行为(图3c)。R(3.09→1.34)和(SPr)2V(1.56→0.68)在浓度增加时表现出明显酸化,表明酸性盐解离。相比之下,[(NPr)2V]Cl4的pH值几乎保持不变,与非水解季铵盐电荷稳定相一致。pH趋势的差异与官能团质子解离热力学直接相关,揭示了以下解离能力顺序:-SO3−>-OH > -N(Me)3+。
随后,作者团队通过CV分析,系统性pH变化滴定揭示了三种紫精衍生物在碱性条件下的明显耐受性差异。当pH从4.7升至13时,R表现出稳定的氧化还原行为,其E1/2从−0.37 V移动至−0.41 V(ΔE= -40 mV),而第二个值保持在-0.76 V(图3d),表明存在质子耦合电子转移(PCET)过程。相比之下,(SPr)2V在碱性条件下均显示出分解特征。对于[(NPr)2V]Cl4,在-0.84 V(pH = 13)处出现非法拉第肩峰,提示部分脱烷基化生成不对称紫精片段(图3e)。(SPr)2V的降解起始更早(pH = 11),以-0.85 V处的不可逆氧化峰为标志,显示出更严重的结构崩解(图3f)。
为阐明pH依赖性结构完整性,作者团队进行了1H NMR研究(pH = 7~13)。R表现出优异的稳定性(pH = 7~11),即使在pH = 11时仍保留质子特征峰(δ:8.62和9.14 ppm),且未出现峰分裂或强度损失(图3g)。相比之下,[(NPr)2V]Cl4和(SPr)2V在pH = 11时发生明显的单烷基化分解。三种紫精盐的质子信号均显示一致的光谱模式(pH > 11):右侧质子的强度和积分明显超过左侧质子,表明从单侧烷基化降解转变为双侧烷基化降解(图3h,i)。此外,解离能测量表明,R–异构体(352.9894 kJ mol−1)相较于[(NPr)2V]Cl4(315.3397 kJ mol−1)和(SPr)2V(310.7185 kJ mol−1)具有明显更高的能垒,表明R构型具有更优的键稳定化(图3j)。
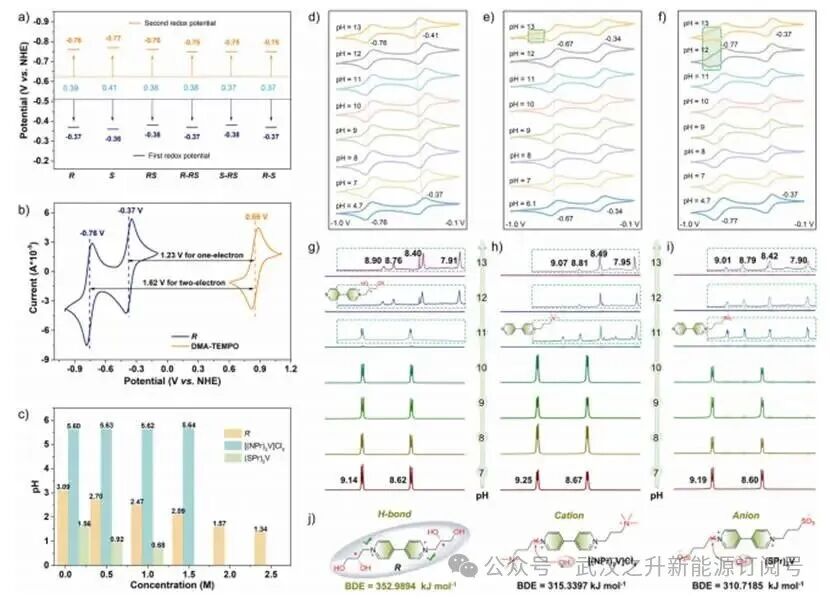
图3. a) 手性紫精氧化还原电位的统计分析;b)化合物R和DMAP-TEMPO在0.1 V s−1条件下的循环伏安图;c)三种紫精衍生物(R、[(NPr)2V]Cl4和(SPr)2V在不同浓度条件下的pH变化;d)R、e)[(NPr)2V]Cl4和f)(SPr)2V在不同pH条件下的CV曲线变化;g)R、h) [(NPr)2V]Cl4和i)(SPr)2V在不同pH条件下的1H NMR谱变化;j)三种紫精衍生物的键解离能(BDE)计算。
5. 分子动力学模拟
分子动力学(MD)模拟显示,R保持均质分散。定量分析表明,溶质-溶剂氢键(OH···H2O:400个计数)明显超过分子内-OH自缔合(8个计数),确立了溶质-溶剂相互作用主导地位作为R优异溶解度的主要决定因素(图4a,b)。残余OH···Cl氢键(46个计数)源于极化离子-偶极相互作用。-N(Me)3+因三甲基取代导致空间位阻诱导的氢键缺陷,使得溶解过程主要由弱离子键解离主导。相反,-SO3−在溶剂化壳层内组装出强效协同氢键网络(740个计数),其多重电负性氧位点作为氢键受体介导该过程(图4c)。径向分布函数(RDF)分析阐明,i通过-OH基团实现双向配位,同时作为氢键供体和受体。OH···H2O的第一溶剂化壳层显示配位数为4(r = 2.4 Å,N = 1),在次级壳层中增至15个协同水(r = 3.8 Å;N = 3.8)(图4d)。此外,OH···Cl相互作用显示出明显降低的N值为0.16(r = 2.8 Å)。结果表明,在R的溶剂化过程中,水优先占据邻近配位位点,而Cl−倾向于形成弱外围相互作用。在较远的径向距离(2.8 Å,r > 2.4 Å)处可观察到渐进的水合层结构,水合层共同构建了溶剂化保护层,是二羟基手性紫精碱类化合物耐碱性的关键决定因素。相比之下,[(NPr)2V]Cl4表现出可忽略的氢键参与度,其阳离子铵核周围由静电作用聚集的Cl−构成。虽然(SPr)2V通过S=O键(N = 1.7,r = 2.4 Å)展现出强氢键接受能力,但溶剂化效应仅局限于末端基团,导致紫精碱骨架完全暴露于溶剂中(图4e,f),从而加速了碱介导的脱烷基化降解过程。
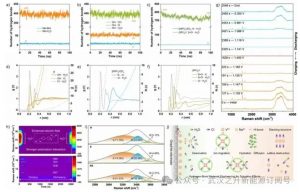
图4. 1.0 M R在1.0 M NaCl中的MD模拟快照,时间分别为0 ns(左)和100 ns(右);时间依赖性氢键计数:a) 分子内(Mol-Mol)和溶质–水(Mol-H2O),Mol代表R;b) OH···OH、OH···Cl和OH···H2O;c) [(NPr)2V]Cl4的N···H2O和(SPr)2V的S=O···H2O;d) R、e) [(NPr)2V]Cl4和f) (SPr)2V的配位数RDF;10 mM R的原位电化学–拉曼光谱表征:g) 二维光谱轮廓和h) 三维光谱映射;i) 1 M手性紫精对H2O···O─H拉伸模式峰值频率的影响;j) R的可能溶解–溶剂化机制示意图。
6. 氢键网络测试
原位拉曼光谱被用于监测施加电位变化时R–紫精的特征峰演变(图4g,h)。在充放电过程中,3000~3800 cm−1处的水特征峰强度明显衰减,1.4V时完全消失。后续放电导致水峰逐渐再生,主要源于界面水的重新取向(“H-down”构型)和解离,以及电场增强下氢键网络的重构。这些变化通过降低界面浓度和局部屏蔽效应共同减弱了水的拉曼信号。在O-H伸缩振动区(2700~3800 cm−1),异构体R/S与外消旋体RS之间观察到明显的振动谱差异。峰解卷积揭示了三种不同的氢键模式:强、中和弱。定量分析表明,R和S分别将71.79%和74.38%的积分强度分配给强氢键区域,而中等/弱贡献分别为23.09%/5.12%(R)和20.96%/4.66%(S)(图4i)。相比之下,RS表现出增强的强氢键贡献(81.53%)以及减弱的中/弱贡献(16.16%/2.31%),揭示了一种协同机制:手性构型同时削弱了水分子间的氢键作用,同时增强了紫精-H2O的相互作用。
基于上述发现,作者团队提出了分子R的全面溶解–溶剂化机制:固态堆积晶格在能量吸收作用下解体为离散分子物种,这些分子扩散进入溶剂介质(图4j)。随后的水相重构通过羟基–水配位建立双向氢键溶剂化结构,同步离子迁移过程稳定了保护性水合层。在电极附近区域,该溶剂化壳层通过场调制去屏蔽效应响应电双层微环境,从而实现高效电子转移。氢键效应不仅加速了溶解动力学,还通过溶剂化结构的热力学稳定抑制了副反应。
7. 液流电池研究
基于0.1M R/DMAP-TEMPO的AORFB进行的解耦电池测试显示,该材料具有优异的单电子循环稳定性,同时具备亚稳态双电子存储能力(图5a)。系统电压测试(0.1~2.3 V)表明,在双电子还原过程中(R2+→R0)存在双重充放电曲线,可实现5 Ah L−1的峰值容量。然而,快速衰减现象(30次循环内损耗20%)提示其存在不稳定性,需对单个氧化还原步骤进行解耦分析。在首个氧化还原窗口内(0.1~1.6 V,R2+→R+•)进行受控循环时,材料展现出优异稳定性(100次循环内容量保持率100%,初始容量2.2 Ah L−1)。第二个过程(1.6~2.3 V,R+•→R)则呈现快速衰减(30次循环内容量从1.9 Ah L−1降至1.5 Ah L−1),证实了R0物种(不溶性)的固有不稳定性。降解机制源于相分离行为,从根本上限制了水介质中深度还原态的电化学可逆性。
在运行窗口(-0.4~0.4 V)下,作者团队采用1 M R2+/R+•氧化还原电对的对称电池在3652次循环(57天)内表现出99.42%的容量衰减,是迄今为止报道的基于紫精电解质中循环稳定性最高的(图5b)。基于1 M R/DMAP-TEMPO的AORFB在长时间循环(533次循环)中表现出100%的容量保留,优于采用1 M [(NPr)2V]Cl4/DMAP-TEMPO(95.6%)和(SPr)2V/K4Fe(CN)6(65.49%)的对应物(图5c)。
进一步的电池性能评估表明,其他基于手性紫精的AORFBs在1 M浓度下经过100次恒电流充放电循环后,容量保持率表现出明显一致性,具体保留率分别为99.94%、99.42%、99.81%、99.59%和99.37%(图5e),有力证实氢键介导的溶剂化保护机制能有效增强分子构象的稳定性。在60~200 mA cm−2电流密度梯度下进行恒电流循环测试时,整个测试区间均保持稳定的库仑效率(CE = 99.9%)(图5f)。1 M R/DAMP-TEMPO基AORFB的能量效率(EE)和电压效率(VE)随电流密度增加呈现渐进式衰减,分别从81.6%降至50.9%(EE),放电容量从11.55降至9.38 Ah L−1。这种衰减归因于高电流密度下欧姆极化效应的增强,导致明显的电压降及效率下降。极化分析显示其具有优异的功率密度特性:在384 mA cm−2(100% SOC)时达到238 mW cm−2峰值输出,在354 mA cm−2(50% SOC)时为202 mW cm−2(图5g)。
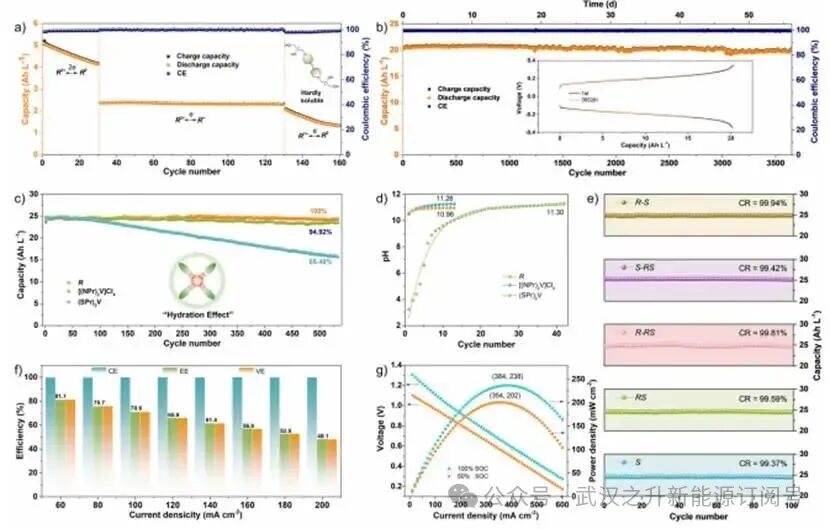
图5. a) 0.1 M R/DMAP-TEMPO基AORFB 在40 mA cm−2下的长期循环性能;b) 1 M R2+/R+•在40 mA cm−2下的对称电池性能;c) 三种紫精体系的循环稳定性比较(533次循环,60 mA cm−2):R/DMAP-TEMPO、[(NPr)2V]Cl4/[(NPr)2V]Cl4和(SPr)2V/K4Fe(CN)6基AORFB;d) 三种紫精在0.1 M溶液中的原位pH演变;e) 1 M S-、RS-、R-RS、S-RS和R-S/DMAP-TEMPO基AORFBs在60 mA cm−2下100次循环的长期循环性能;f) 1 M R/DMAP-TEMPO基AORFB在20 mA cm−2增量下从60到200 mA cm−2的库仑效率(CE)、能量效率(EE)和电压效率(VE);g) 1 M R/DMAP-TEMPO基AORFB在10 mA cm−2下充满电后的极化行为和功率密度特性
8. 高浓度电池性能测试
在兆瓦时级RFBs中,电解液成本占能量组分总成本的40%,体积与电解液浓度呈反比关系。成本–浓度的相互依赖关系使得高浓度耐受性成为实际应用的关键决定因素。为评估分子R的经济可行性,作者团队设计了浓度梯度AORFB系统(1.5、2.0和2.5 M)。而且,基于1.5 M R/DMAP-TEMPO的AORFB在595次连续循环中保持100%容量保留率,而2.0 M高浓度电池在453次循环中保持99.98%容量保留率(日变化率0.0003%),放电容量达46.60 Ah L−1(图6a,b)。突破浓度极限后,2.5 M R/DMAP-TEMPO的AORFB达到67 Ah L−1理论容量,125次循环保持100%容量保留率,在88.97%利用率下实现59.61 Ah L−1实际容量(能量效率68.5%,电化学效率99.9%)(图6a,b)。与传统浓度依赖性衰减模式相反,异常稳定性梯度源于二羟基手性紫精的独特溶剂化结构调控。然而,高浓度下逐渐出现的容量衰减源于渗透压驱动的水分子渗透。
9. 放大技术的验证
作者团队通过自主研发的20 L常压反应系统实现了R材料的规模化公斤级合成(图6c)。在2 M电解液浓度条件下,采用五组单电池串联电堆结构进行放大性能评估。该系统在77次循环中保持稳定运行,放电容量达13.41 Ah,容量利用率为83.40%,容量保留率为98.65%(图6d)。平均充放电电压(7.206 V和3.767 V)实现52.28%的VE。本研究首次成功展示公斤–安时级中性AORFB,可为LED灯、微型风扇等终端设备提供动力。多维度性能测试表明,手性紫精R在关键电池参数上具有明显优势(图6e、f)
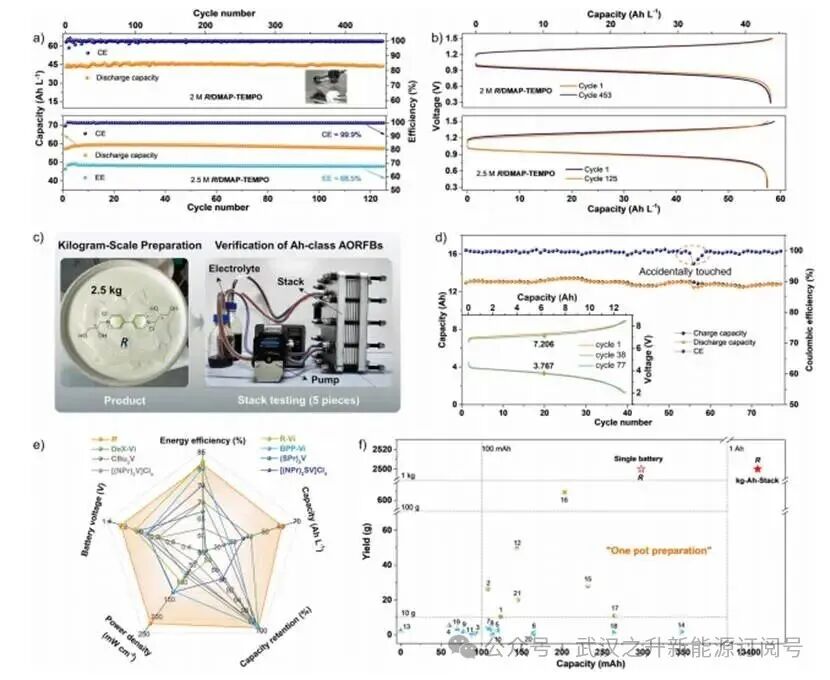
图6. a) 2 M(上)和2.5 M(下)R/DMAP-TEMPO的AORFB在60 mA/cm²电流密度下的长期循环性能;b) 对应的充放电曲线;c) 千克级R制备与堆叠级测试流程;d) 负极/正极电解液浓度为2 M/1.2 M、容量为300 mL/500 mL的Ah级电池性能评估;e) 紫精材料综合性能对比雷达图;f) 紫精基中性AORFBs性能对比
结论展望
本研究整合了一个基于大型语言模型(LLM)的机器学习框架,以指导手性紫精电解质的合理设计和合成,开创了一种手性调控、氢键介导的溶剂化工程策略,用于高性能中性水系有机液流电池(AORFBs)。通过分析超过1300项AORFB研究的大型数据集,机器学习模型能够预测影响电池稳定性和性能的分子特征,从而指导具有定制化溶剂化结构的紫精衍生物的合成。溶解度测试显示,单一对映体R和S分别实现了2.75 M和2.76 M的明显水溶性,较外消旋混合物(1.66 M)提高了1.66倍,归因于邻位二羟基取代基之间立体定向的分子内氢键作用,该作用通过重组溶剂化环境形成动态的“溶剂化装甲”。结合原位光谱表征的整合分子动力学模拟阐明了这种氢键网络稳定分子结构的机制,有效屏蔽了吡啶鎓C-N键在pH = 11范围内免受氢氧根离子亲核攻击。由此产生的pH适应性溶剂化防护层赋予了优异的碱性耐受性,转化为优异的电化学稳定性。通过R2+/R+•在对称电池中展现出3652次循环(57天)后仍保持99.42%的容量,创下有机负极电解液中最高的记录稳定性。将手性紫精与DMAP-TEMPO组装后,可实现533次循环的100%容量保留率,优于阳离子[(NPr)2V]Cl4(94.92%)和阴离子(SPr)2V(65.49%)电解质。在实验室规模验证基础上,作者团队成功实现了优化手性紫精的公斤级合成(2.5公斤批次),证明了合成可扩展性。安时级电池电堆测试进一步证实了电解质的实际应用性,在相关运行条件下,经过77次循环后,容量保持率仍高达98.65%。本研究通过将先进机器学习与立体化学溶剂化工程及可扩展合成技术相结合,为数据驱动型电解质设计开创了全新范式,不仅在分子稳定性和电化学性能方面取得突破性进展,更成功弥合了中性AORFBs实现工业化应用的鸿沟。
文献信息
Xu Liu, Haiyan Yu, Xiaotong Deng, Jian-Yue He, Xuri Zhang, Junjie Huang, Zengrong Wang, Chenjing Liu, Xin Zhang, and Gang He, Machine Learning–Guided Solvation Engineering of Chiral Viologens for Durable Neutral Aqueous Organic Flow Batteries, 2025, Angewandte Chemie International Edition
doi.org/10.1002/anie.202522442