鉴于铬离子反应动力学缓慢限制了铁铬液流电池(ICRFB)的性能,中科院过程工程研究所张洋团队以盐酸胍作为负极电解液的添加剂以提高能量效率并抑制析氢反应。含有0.4 M盐酸胍的电解液显示出最低的过电位,在55°C下达到80%的最高能量效率,比原始电解液高出6%。一方面盐酸胍提高了铬离子的反应性,另一方面盐酸胍的存在增加了铬离子第一水合层中Cl–的数量,从而提高了铬离子还原反应的活性。此外,原位拉曼光谱表明当加入盐酸胍时,电极附近水的氢键网络被破坏,氢离子的转移受到阻碍,抑制了析氢反应。因此,盐酸胍增强了铬离子的反应性,提高了ICRFB的性能。相关成果以“Enhancing Battery Performance through Solvation StructureModulation of Iron−Chromium Electrolytes Using Guanidine Hydrochloride”为题发表在ACS Applied Energy Materials上。感谢中科院过程工程研究所李兆昕(第一作者)校稿!
本文所用
一体化液流单电池测试系统(YTH-1/LSB-1)
由武汉之升新能源有限公司提供

汇聚液流电池科研人员超1200人
长按识别下方二维码,邀请进群
(备注:单位名称姓名电话、进群)
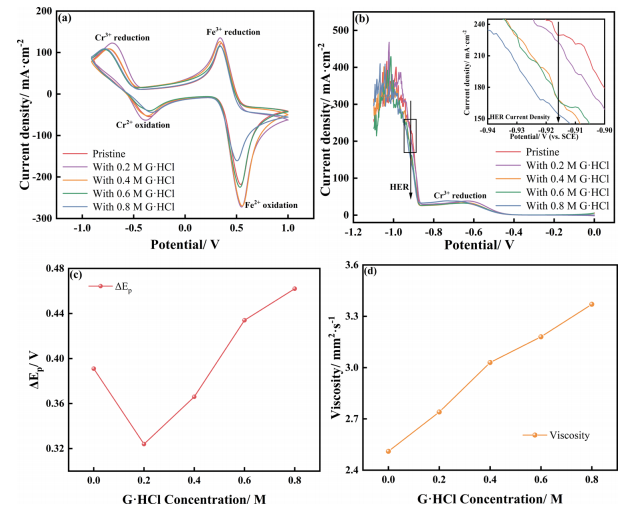
通常,小分子络合剂形成络合物的能力较弱,尤其是在酸性电解液中。在ICRFBs中,尽管在如此强的酸性溶液中形成稳定的络合物具有挑战性,但可以通过使用添加剂来影响溶剂化环境并改变离子行为。因此,应引入分子量低但具有足够供电子官能团(如氨基)的络合剂,以确保影响铬离子的能力。盐酸胍(G·HCl)因其“三个强路易斯碱基共面氨基”而成为理想的添加剂。在水溶液中,GH+离子的三个键长和键角几乎相等,电子表现出“Y–离域”。通过这种结构,分子的共振稳定使正电荷均匀地分布在三个氮原子周围,减少了对阴离子的吸引力。这种溶液环境允许Cl–离子与铬离子发生更多的相互作用,使铬离子更容易获得电子。因此,G·HCl有望影响铬离子的水合结构,提高其氧化还原性能。在这项研究中,为了提高ICRF的能量效率并抑制HER,在电解液中加入了G·HCl。首先,进行了电化学测试和电池测试以直接显示G·HCl的作用。之后,电化学阻抗谱(EIS)和塔菲尔曲线显示了G·HCl对动力学的影响。最后,通过光谱技术和分子动力学模拟研究了G·HCl的机理。通过RDF曲线研究了G·HCl对铬离子溶剂化环境的影响,并解释了铬离子反应性的提高,相关结果得到了紫外–可见光谱的验证。原位拉曼光谱用于探索抑制HER的机制。 图1a显示,0.2 M G·HCl电解液中铬的氧化还原峰表现出最佳对称性。如图1c所示,ΔEp最初随着G·HCl浓度的增加而减小,然后增加。当G·HCl浓度为0.2 M时,ΔEp达到最小值0.324 V,表明Cr3+/Cr2+的可逆性最佳。当G·HCl的浓度从0.2 M增加到0.8 M时,ΔEp从0.324 V增加到0.461 V,电化学可逆性的降低可能是由于电解液粘度的增加造成的,如图1d所示。还可以注意到G·HCl的存在也会影响Fe3+/Fe2+氧化还原电对。然而,当向阴极电解液中加入G·HCl时,电池在放电过程中的过电位会增加。原因可能是添加G·HCl会增加电解液的粘度(图1d)并降低传质效率。图1b显示了不同浓度G·HCl的电解液的LSV曲线。当电势达到-0.6 V(vs SCE)时,三价铬离子开始被还原,并观察到平滑的电流峰值。随着扫描电压继续降低(-0.9 V),HER开始出现,导致电流突然跳变。垂直的黑色箭头表明G·HCl抑制了HER的电流密度。在-0.915 V的电压下,原始电解液的电流密度为230 mA·cm−2,而含有0.8 M G·HCl的电解液电流密度仅为152 mA·cm-2。图1b还显示,当G·HCl添加到0.4 M时,效果明显优于原始电解液,电流密度为165 mA·cm−2。最后,当电压降至约-0.95V时,工作电极上会不断产生H2,曲线会振荡。
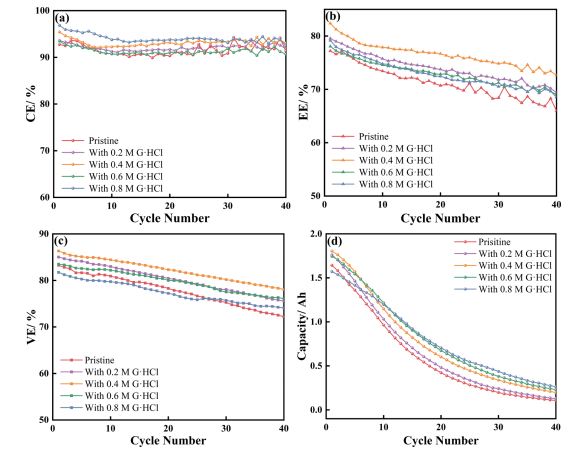
图1 含0、0.2、0.4、0.6和0.8 M G.HCl的原始电解液的(a) CV和(b) LSV曲线;(c)Cr3+/Cr2+的峰值电位差(ΔEp)和电解液的(d)粘度随G·HCl浓度的函数如图2a所示,G·HCl对CE的增强没有显示出明显的规律性,可能是因为CE取决于许多因素,如HER和金属离子交叉。如图2b所示,与原始电解液相比,0.4 M G·HCl的电池的能量效率超过80%,提高了6%。图2c是VE曲线,更准确地反映了G·HCl反应性的提高。随着G·HCl浓度的增加,VE先增加后减少,在0.4 M时达到最大值87%,这是最佳浓度。然而,图1a显示,含有0.2M G·HCl的电解液表现出最佳铬氧化还原可逆性,这种差异可归因于扩散和动力学引起的过电位。
图2 (a)库仑效率(CE)、(b)能量效率(EE)和(c)电压效率(VE)作为不同浓度G·HCl条件下电解液循环数的函数,CE数据在30次循环后由于电池容量较低而出现波动;(d)电荷容量与0、0.2、0.4、0.6和0.8 M的原始电解液循环数的函数图2d显示含0.8MG·HCl的电解液在30个循环内的平均衰减率为2.40%,明显低于在原始电解液中观察到的2.93%。经过20个循环后,0.8M G·HCl电解液的容量是原始电解液的1.67倍,说明G·HCl对容量衰减的抑制作用。然而,含0.8MG·HCl的电解液的初始容量仅为原始电解液的1.17倍。结果表明G·HCl减轻了容量的衰减。
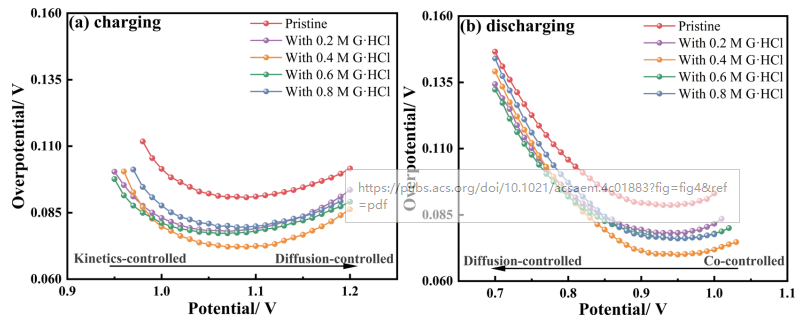
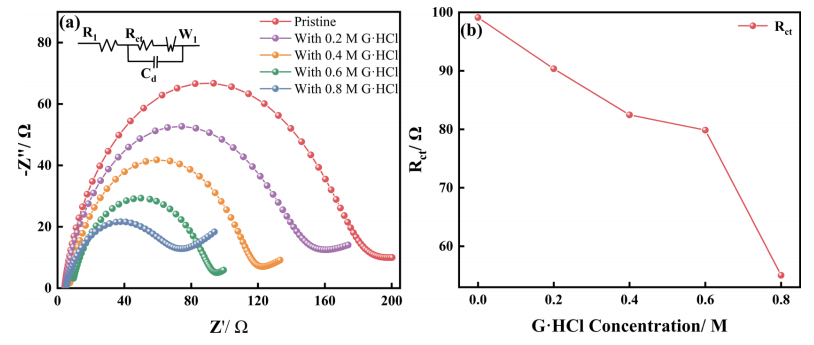
图3 不同浓度G·HCl的电解液中(a)充电和(b)放电随工作电位变化的过电位曲线如图3a所示,在充电过程开始时,Cr3+的缓慢反应活性需要更高的电化学过电位来引发反应,原始电解液的过电位为0.112 V。随着充电的进行,反应物浓度的降低增加了扩散的重要性。因此,极化电压增加。当加入0.4 M G·HCl时,电解液表现出动力学和反应物扩散的良好结合,在大多数潜在条件下,过电位保持在最低值。对于放电过程(图3b),过电位在开始时保持在一个稳定的低值(1.0−0.9V)。在这一过程中,Cr2+极容易氧化。因此,不需要高过电位来驱动反应,反应速率受动力学和扩散的控制。在这一阶段,含0.4MG·HCl的电解液的性能最好,在0.92 V的电压下,过电位为0.070 V。与0.089 V的原始电解液相比,过电位降低了约19.1%。在放电过程结束时,电极表面的反应速率受到传质过程的控制。因此,G·HCl增强反应动力学的能力明显降低,导致原始电解液与0.4M G·HCl电解液之间的过电位差小于0.008 V。充放电过程均表明在加入0.4M G·HCl后,过电位达到其最小值。因此,0.4 M是G.HCl的最佳浓度。 EIS的等效电路如图4a。所示随着G·HCl浓度的增加,奈奎斯特图中的高频弧的半径减小,与图4b所示的Rct的趋势相对应。与图1c中所示的ΔEp不同,Rct的测量不受粘度的影响。因此,Rct随G·HCl浓度的变化趋势是一致的,并且不像ΔEp那样波动。Rct的降低表明电荷转移过程的能垒较低,并且G·HCl对ICRFB性能的提高可以归因于铬离子氧化还原反应动力学的改善。
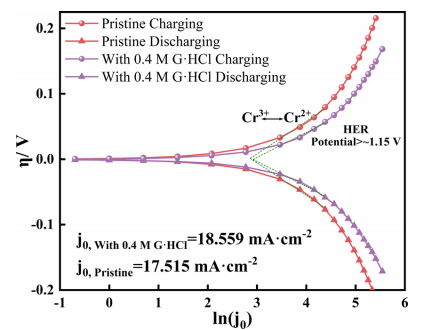
交换电流密度(j0)是评价电化学反应活性的关键指标。进一步研究G·HCl如何影响Cr3+/Cr2+电子转移反应性,原始电解液曲线的电解液和含0.4MG·HCl的电解液被用来测量交换电流密度(j0)。在电流密度大于100mA·cm−2(ln j0>4.6)的区域,电势大于1.1V,伴随着HER。选择电流密度在32至80 mA·cm−2范围内的区域进行线性拟合,因为这个范围对应于涉及铬氧化的主要反应。充电线和放电线交叉处的电流密度为j0。图5显示与原始电解液相比,含有0.4 M G·HCl的电解液的过电位较低。前者的j0比后者高1.044mA·cm-2(约6%)。根据Butler-Volmer模型,更高的j0意味着正极和负极的反应性更好。因此,添加G·HCl改善了铬氧化还原反应的动力学。
图5.在电池中加入0.4MG·HCl的原始电解液和原始电解液的Tafel曲线
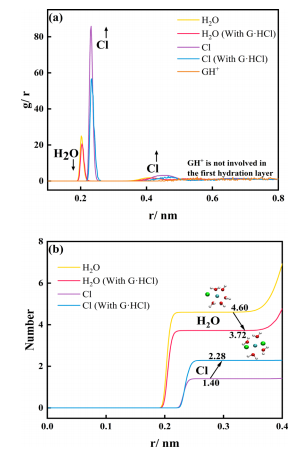
图6 含和不含0.4M G·HCl的电解液中铬离子的(a) RDF曲线;(b)计算了含/不含0.4 MG·HCl的电解液中铬离子的第一水合层的组成图6a,b显示在含有0.4MG·HCl的电解液中铬离子的第一个水化层平均含有2.28Cl−,高于原始电解液中的1.40。此外,G·HCl并不侵入铬离子的第一水化层,而是减少了水分子的平均数量,增加了Cl−的数量。考虑到Cr(H2O)63+的反应性较差,其中较高浓度的G·HCl导致Cr3+/Cr2+的Rct较低,可以推断Cl−促进了电子转移反应。为了验证上述计算结果,进行了紫外−可见光谱测试。首先,对不同老化状态下的电解液进行了测试,以显示电解液活性与光谱测试结果之间的关系。然后,对含0.4M G·HCl的电解液进行了测试,研究了其对铬离子附近Cl−数的影响效果。对含有0.4 M氯化钾的电解液进行了测试,以消除观察结果是由Cl−浓度增加引起的可能性。
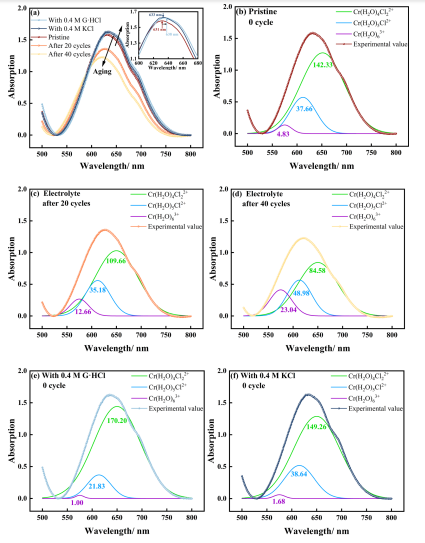
图7 不同老化状态的电解液、0.4MG·HCl电解液和0.4 M氯化钾电解液的(a)紫外−可见光谱分析;[Cr(H2O)4Cl2]+,[Cr(H2O)5Cl]2+,[Cr(H2O)6]3+(b)原始电解液,原始电解液经历(c) 20循环和(d) 40循环,(e)电解液与0.4g·G HCl和(f)电解液+0.4氯化钾的分离峰图7a显示随着循环次数的增加,峰值显示蓝移(从631到621 nm),而添加了G·HCl和KCl的电解液的峰值显示红移(从631-638 nm)。尽管KCl和G·HCl电解液的曲线看起来非常相似,但两条曲线的几何接近度受到吸光度变化的影响,而峰位的红移最准确地反映了离子种类的变化。添加KCl的电解液峰值(633nm)接近原始电解液,表明KCl对Cr3+水合层的影响有限。图7b−d显示随着老化过程的进行,[Cr(H2O)6]3+的峰面积从4.83增加到23.04,[Cl(H2O)4Cl2]+从142.33减少到84.58。图7e、f显示KCl和G·HCl都能提高[Cr(H2O)4Cl2]+的浓度。其中,G·HCl使峰面积从142.33增加到170.20,对提高铬离子第一水合层中Cl–的数量效果最佳。图7f显示通过KCl引入相同浓度Cl–的电解液的紫外吸收仅149.26,表明这种效应与胍有关,而不仅仅是由于Cl–的增加。然而,图6b表明铬离子第一水合层中的Cl–数量可能>2。虽然不能排除在波长大于650nm处出现新物质吸收峰的可能性,但将其归因于[Cr(H2O)4Cl2]+的峰并不影响G·HCl效应的观察结果。
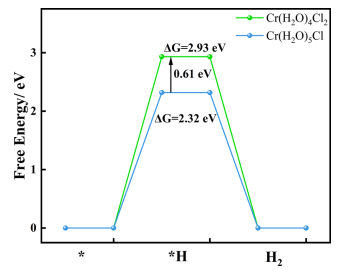
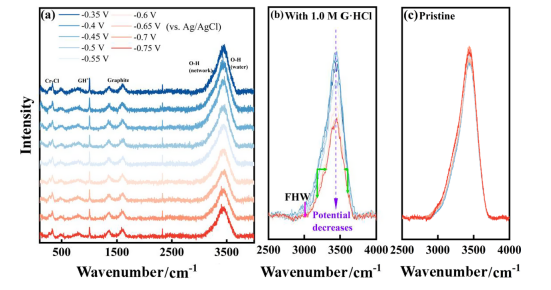
图8.计算了电解液中存在两种不同铬离子的HER的能垒从图8可以看出在铬离子附近水分子较少的溶液环境中,H+在石墨电极上的吸附反应表现出较高的能垒,因此HER受到抑制。如图2d所示,G·HCl对析氢反应的抑制作用的实验结果可以支持计算结果。为了进一步研究G·HCl的作用机理,采用原位拉曼光谱法研究了石墨上的电极反应。对原始电解液和1.0MG·HCl的电解液进行了原位拉曼光谱分析,研究了G·HCl对析氢过程的影响。两种电解液中水的拉曼位移的结果和比较如图9所示。
图9(a)在-0.35至-0.75 V(相对于Ag/AgCl)的电势下,含有1.0 M G·HCl的电解液的拉曼光谱;在2500至4000 cm-1的不同电势下,(b)1.0 M G·HCl和(c)原始电解液中H2O的拉曼位移根据之前的研究,3000至3700 cm-1之间的宽范围拉曼位移结合了具有不同氢键结构的水分子的拉曼位移。其中,约3250 cm-1处的峰值归因于“完全氢键水(FHW)”,而3300至3600 cm-1范围内的峰值则归因于“部分氢键水(PHW)”。自由水(FW)的拉曼位移位于3650 cm−1。图9a、b显示水分子中O-H振动引起的拉曼位移在-0.35至-0.45 V的电位范围内保持相对稳定。当电位降至-0.5 V时,与图1所示的Cr3+还原电位一致,拉曼位移的强度开始降低。图9b中的绿色箭头显示强度在3200 cm-1处显著降低,同时该位置有明显的蓝移,大于3650 cm-1处的红移。品红色箭头显示位于3000–3100 cm-1附近的拉曼位移与高度氢键化的水分子有关,随着电位的降低,拉曼位移逐渐降低到基线水平,表明氢键结构被破坏。当电势降至-0.65 V时,Cr3+的还原被过电势加速,这种拉曼位移的强度继续减弱。当达到HER的电势(-0.7至-0.75 V)时,这一趋势终止,拉曼位移不再变化。然而,图9c显示原始电解液的拉曼光谱没有表现出相同的现象。原始电解液与1.0M G·HCl电解液的拉曼光谱差异反映在Cr3+还原反应阶段,说明在电解液中加入G·HCl仅在反应过程中破坏了水的氢键网络本研究表明G·HCl可以提高电解液中铬离子的反应性。在ICRFB中,在负电解液中加入0.4M G·HCl可提高能量效率6%,并抑制HER。此外,含0.4M G·HCl的电解液经过20个循环后的剩余容量超过了原始电解液,其值为46.0%。G·HCl的存在增加了铬离子第一水化层中Cl−离子的数量,从而提高了其反应活性。被较多Cl−离子包围的铬离子对H+离子具有较高的吸收能,并有效抑制HER。此外,Cl−离子的存在破坏了电极附近水分子的氢键网络,阻碍了H+离子的转移,进一步抑制了HER。 Zhaoxin Li, Yang Zhang, Shili Zheng, Huayi Tan, Yihan Deng, Jiuchuan Liu, and Bingqiang Fan,Enhancing Battery Performance through Solvation Structure Modulation of Iron−Chromium Electrolytes Using Guanidine Hydrochloride.2024,ACS Applied Energy Materials
https://doi.org/10.1021/acsaem.4c01883