
第一作者:牛峥嵘
通讯作者:冯海涛
通讯单位:中国科学院青海盐湖研究所
DOI:10.1016/j.electacta.2026.149301

本研究证明乙酸铵(AMA)可作为单向分子开关,通过逐步调控Cr³⁺配位微环境来触发不可逆配体交换催化:NH₄⁺首先促使配位H₂O被Cl⁻取代,从而“激活”电活性中间体[Cr(H₂O)₅Cl]²⁺;随后乙酸阴离子(Ac⁻)置换Cl⁻,使高扩散性的[Cr(H₂O)₅(Ac)]²⁺配合物“稳定”,完成单向配体交换催化过程并增强Cr³⁺扩散。此机制使电池循环寿命延长近600倍(1000次循环后容量保持率达36%,而原始电解液在100次循环后仅保持0.06%),同时将HER过电位提升60mV,在40mA cm⁻²下实现稳定的345mAh放电容量。
《2025年我司用户发表的液流电池论文合集》
铁铬液流电池(ICRFBs)凭借卓越的成本竞争力、丰富的元素组成及良好的环境兼容性,引发了广泛的研究关注,成为电网级储能领域极具前景的解决方案。然而,在实际应用中,ICRFBs的性能会因常温运行条件下固有的电解质失活现象而显著受限。在酸性水介质中,Cr³⁺主要以动力学惰性的d²sp³杂化配位物种形式存在,其中八面体对称结构的[Cr(H₂O)₆]³⁺配合物具有最高的热力学稳定性。这种闭壳层配位结构赋予其极强的动力学惰性,导致电极界面处的电化学氧化还原反应速率极为缓慢。动力学效率低下不仅严重抑制了Cr3+/Cr2+氧化还原反应动力学,而且在负极引起了明显的HER寄生副反应,伴随着电极–电解质界面局部pH升高。电化学惰性物种[Cr(H2O)6]3+与电活性物种[Cr(H2O)5Cl]2+之间的化学平衡是决定电解质活性的关键因素,然而过去二十年间对铬配位络合物生成过程的深入微观研究仍较为匮乏。为突破此关键瓶颈,科研界投入大量精力通过添加多种电解质添加剂来改善Cr3+/Cr2+的氧化还原动力学并抑制HER。
1.电解质电化学特性的优化
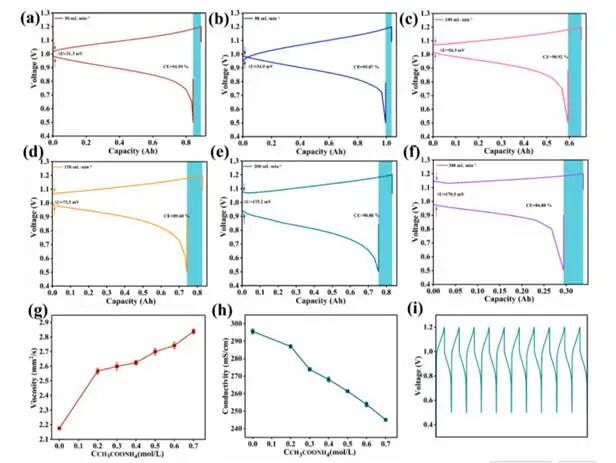
图1电解液优化
在50mL min⁻¹时,系统实现了最小的电压滞后(ΔU=31.3mV)和优异的库仑效率(CE=94.59%)。充足的电解液停留时间可确保Cr3+/Cr2+氧化还原反应充分进行,同时避免因流体剪切力过大导致的界面扰动。将流速提升至80mL min-1时,CE提高至95.87%,极化值也略微增大(ΔU=34.9mV)。当流速进一步从100mLmin-1增至300mLmin-1时,CE逐渐下降并伴随极化持续增强:CE依次降至90.92%、89.68%、90.88%和86.88%,而ΔU则连续上升至54.3mV、73.5mV、133.2mV和170.5mV。高流速下的性能衰减主要源于流动不稳定性、物质停留时间缩短以及泵送损耗增加。最终,确定所有实验均采用50mL min-1作为固定流速。
粘度随添加剂浓度增加呈单调上升趋势,从0mol L⁻¹时的2.17mm² s⁻¹升至0.7mol L⁻¹时的2.83mm² s⁻¹,归因于添加剂引起的离子强度增强与溶剂化效应,强化了分子间相互作用并增加了流动阻力。相反,电导率随添加剂浓度的增加而逐渐降低:从0mol L⁻¹时的295mS cm⁻¹降至0.7mol L⁻¹时的245mS cm⁻¹,很可能源于粘度增加对离子迁移率造成的阻碍作用,或离子对/复合物的形成导致自由电荷载流子浓度下降。综上所述,结果表明CH₃COONH₄的引入会在电解质电导率与粘度之间引发显著的权衡关系。
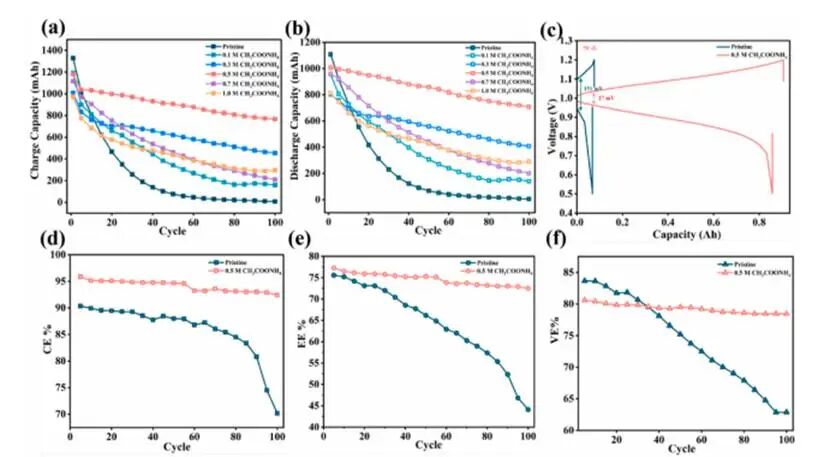
图2使用原始电解液及含有不同浓度乙酸铵的电解液时ICRFBs的电化学性能
含AMA的电解液相较于原始电解液表现出提升的容量保持率,其中0.5M AMA体系最佳。原始电解液呈现渐进且持续的容量衰减现象,其CE、EE和VE均缓慢下降,归因于动力学惰性离子[Cr(H₂O)₆]³⁺的固有失活以及持续存在的HER。与之形成对比的是,经0.5M AMA改性的体系在整个循环过程中始终维持约93%的稳定容量效率、接近73%的高EE以及持续优异的VE。
充放电电压曲线证实在AMA改性体系中极化效应降低,当浓度超过0.5M时,性能会下降,因为过量的AMA会增加电解液粘度并阻碍离子扩散。总体而言,0.5M的AMA浓度在配位活化、质量传输和长期稳定性之间实现了最佳平衡;在此浓度下,电解液在局部配位活化与整体传输阻力之间达到了最优平衡状态。
2.电解质的光谱表征
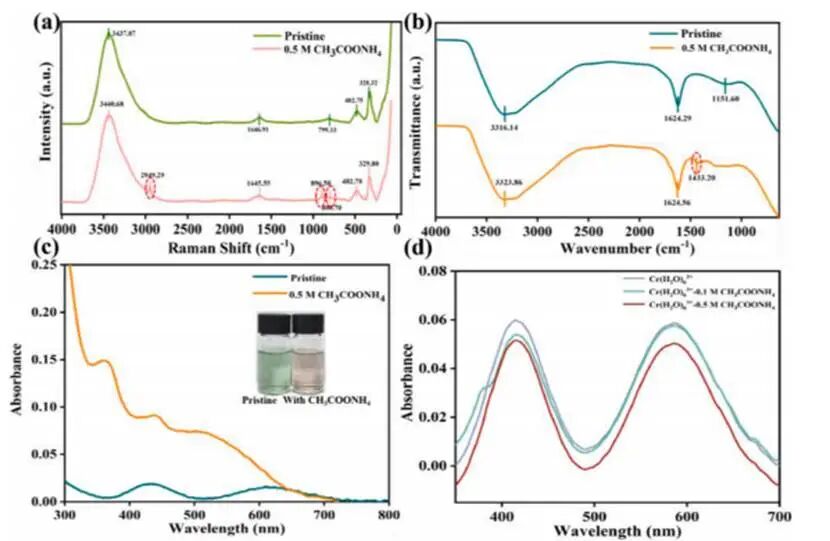
图3添加0.5mol L-1AMA前后电解质配位结构的光谱表征
含有0.5mol L⁻¹AMA的电解液拉曼光谱在2949.29 cm⁻¹和896.50 cm⁻¹处出现两个新的特征峰:2949.29 cm⁻¹处的峰归属于CH₃COO⁻中甲基的C–H伸缩振动;而896.50 cm⁻¹处的峰则源于与乙酸根配位相关的金属–氧(Cr–O/Fe–O)伸缩振动。FT-IR光谱在1433.20 cm⁻¹处显示出一个新的特征吸收峰,对应羧酸根基团(νₛ(COO⁻))的对称伸缩振动。此外,游离AMA中甲基的C–H伸缩峰位于3005–3015 cm⁻¹,而在羧酸根配位后移至2945–2950 cm⁻¹,表明配位壳内分子运动受限导致了约50–60 cm⁻¹的红移。AMA掺入引发了颜色从绿色向粉色的明显转变,并伴随紫外–可见吸收谱的显著变化,表明电解液内部发生了显著的配位结构重排。
3.电化学性能及动力学分析
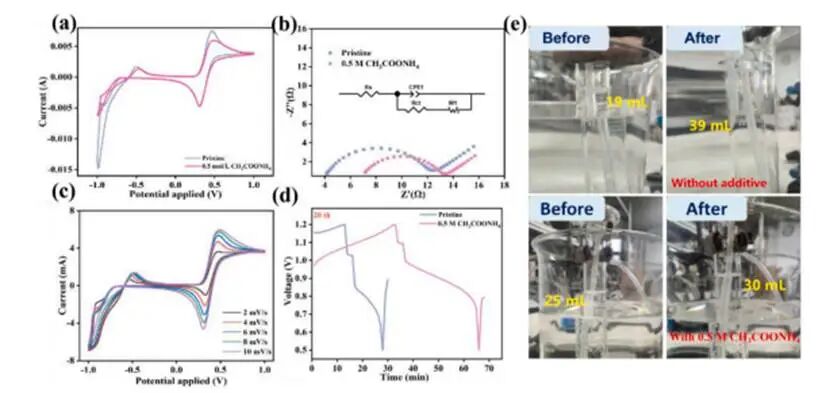
图4电化学动力学分析与气体析出特性表征
含AMA的电解液表现出明显减小的峰电位差,表明氧化还原反应可逆性显著改善。EIS结果显示与原始电解液相比,0.5mol L-1AMA改性电解液展现出更低的串联电阻(Rs:3.3Ω对比4.5Ω)和更低的极化电阻(Rp:8.0Ω对比9.6Ω)。结果证实AMA既提高了电解液的离子导电性,又优化了电极界面处的电荷转移动力学。此外,0.5mol L-1AMA电解液在2至10mV s-1的扫描速率范围内均表现出优异的CV稳定性,而原始电解液则不然。原始电解液存在严重的电压极化现象,而AMA的引入显著抑制了电压损失,从而大幅降低内部极化程度并显著提升电池整体性能。量筒水置换法定量分析显示原始体系在20次循环中产生约20mL气体,而含AMA的体系仅产生5mL气体,相当于氢气释放减少了四倍,证实了AMA的HER抑制作用。
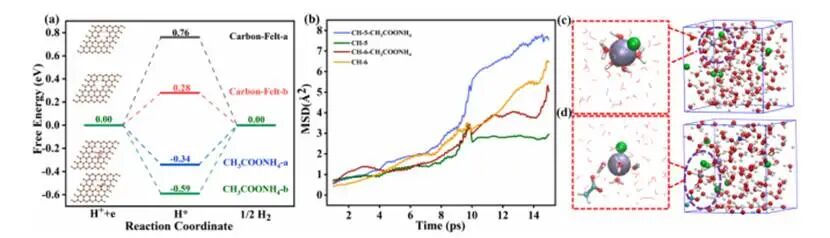
图5.理论计算与离子传输动力学分析
AMA通过稳定电极表面吸附的H*中间体,将HER能垒提升60mV(从280mV增至340mV),并将速率决定步骤从H*吸附转变为H*脱附,从而有效抑制副产物氢气的生成。不同电解质体系的均方位移分析显示在15ps时间点,[Cr(H₂O)₅Cl]²⁺(CH-5)与[Cr(H₂O)₆]³⁺(CH-6)的MSD曲线收敛表明两种物种具有相似的整体扩散行为。CH-5-AMA体系展现出卓越的Cr³⁺扩散迁移率,远超其他所有体系;具体而言,不仅能提升Cr³⁺的扩散效率,还能通过使反应过电位升高60mV来抑制HER反应。综上所述,AMA通过调控Cr³⁺的溶剂化结构来加速Cr³⁺/Cr²⁺的氧化还原动力学,从而缓解容量衰减并提升长期稳定性。
4.长期循环使用下的耐久性及电化学稳定性
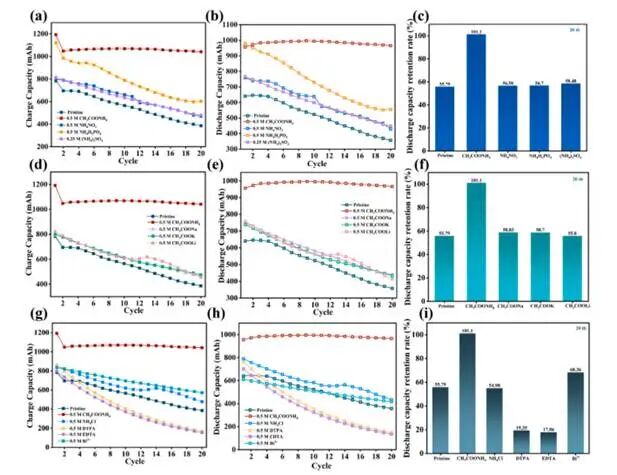
图6.通过系统性控制实验分离NH₄⁺与Ac⁻的贡献,并验证其协同效应
仅CH₃COONH₄在20次循环后实现了101.1%的放电容量保持率。其他铵盐的保持率(56.58%~58.48%)与原始电解液(55.79%)相当,证实了单独使用NH₄⁺无法实现所观察到的稳定性提升。其次,仅CH₃COONH₄达到101.1%的保持率,而其他乙酸盐的保持率(55.8%~58.83%)均接近基线水平,表明单独使用Ac⁻亦不足以提升性能。最后,在相同条件下将AMA与已报道的添加剂(NH₄Cl、Bi³⁺、EDTA、DTPA)进行对比:Bi³⁺仅带来适度改善(68.36%),螯合剂(EDTA、DTPA)则导致性能显著下降(保持率<20%);相比之下,AMA展现出前所未有的稳定性(101.1%保持率),远超其他所有添加剂。综上结果明确表明性能提升源于乙酸铵的协同效应。
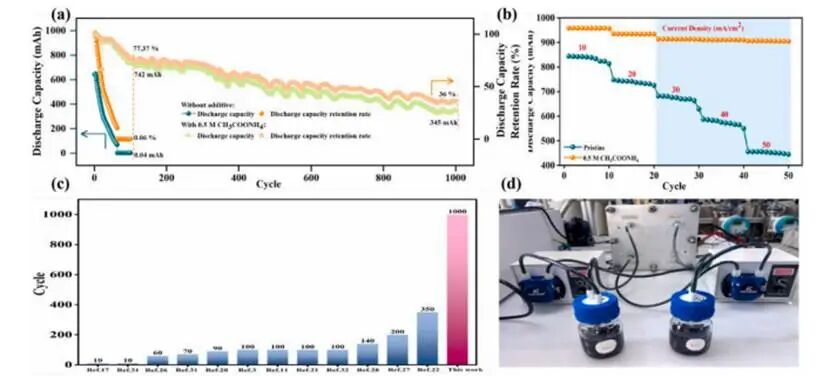
图7.ICRFBs的长期稳定性和倍率性能
经AMA改性的电解质展现出卓越的循环稳定性:经过1000次连续循环后,放电容量仍保持初始值的36%(345mAh),远优于未添加AMA的体系,后者仅需100次循环便出现容量急剧衰减,从640mAh骤降至0.04mAh(容量保留率仅为0.06%)。AMA改性体系不仅具备优异的长期循环性能,其容量保持能力也得到显著提升。电解质的倍率性能显示当电流密度从10mA cm⁻²增至50 mA cm⁻²时,AMA改性电解质的放电容量始终稳定在约900mAh,显著高于原始体系(在30mA cm⁻²下分别为915mAh与690mAh;在50mA cm⁻²下分别为900mAh与410mAh)。
本研究结合系统实验与理论计算揭示了由AMA诱导的不可逆配体交换催化背后单向分子开关机制。通过使用低成本的NH4Ac作为单一组分电解质添加剂,建立了完整的单向配体交换激活路径:NH4+首先驱动不可逆活化反应生成电活性产物[Cr(H2O)6Cl]2+,而乙酸阴离子则通过形成高扩散性的[Cr(H2O)5(Ac)]2+来稳定体系。单向分子开关使CH6→CH5的活化能垒降低17.9kcal mol-1,将HER过电位提升60mV,增强Cr3+扩散速率,并将循环寿命延长近600倍(AMA改性电池在1000次循环后仍保持初始容量的36%,并在40mA cm-2下提供稳定的345mAh放电容量)。