第一作者:房文生、郭巍、陆瑞虎
通讯单位:中国科学技术大学、奥克兰大学、华中科技大学
通讯作者:姚涛、王子运、夏宝玉
【成果简介】
电解将二氧化碳(CO2)减少并转化为有用的化学物质,原则上可以为更可持续和碳中和的未来做出贡献。然而,将其发展成一个强大的过程仍然具有挑战性,因为有效的转化通常需要碱性条件,其中CO2沉淀为碳酸盐,这限制了碳的利用和系统的稳定性。物理洗涤、脉冲操作和使用偶极膜等策略可以部分缓解这些问题,但不能完全解决这些问题。因此,在不形成碳酸盐的酸性电解液中进行二氧化碳电解被认为是一种更可行的解决方案。在这项工作中,作者开发了一种质子交换膜系统,该系统在源自废铅酸电池的催化剂下将CO2还原为甲酸,其中晶格碳活化机制起重要作用。当CO2还原与氢氧化耦合时,甲酸的法拉第效率超过93%。该系统与启动/关闭过程兼容,在电流密度为600 mA cm -2和电池电压为2.2 V的情况下,二氧化碳的单通转换效率接近91%,连续工作时间超过5200小时。系统通过使用强大而高效的催化剂,稳定的三相界面和耐用的膜,实现了卓越的性能,这将有助于推动碳中和技术的发展。
【研究背景】
随着全球经济的增长和社会的快速发展,化石燃料的过度使用导致空气中二氧化碳的浓度逐年递增,从而导致严重的环境污染与气候问题。二氧化碳电解技术为推进双碳战略的实施以及人类社会的可持续发展提供了一种可靠的途径。在诸多产物中,甲酸作为一种重要的液体化学原料,在化工、医药、农业和能源等领域都有着广泛应用。同时,甲酸作为一种重要的氢源媒介,可以大大缓解可再生能源波动性的问题。尽管人们近年来在高性能二氧化碳还原电催化剂取得了进展,但仍面临工况条件材料与系统的服役水平与寿命的重大挑战。
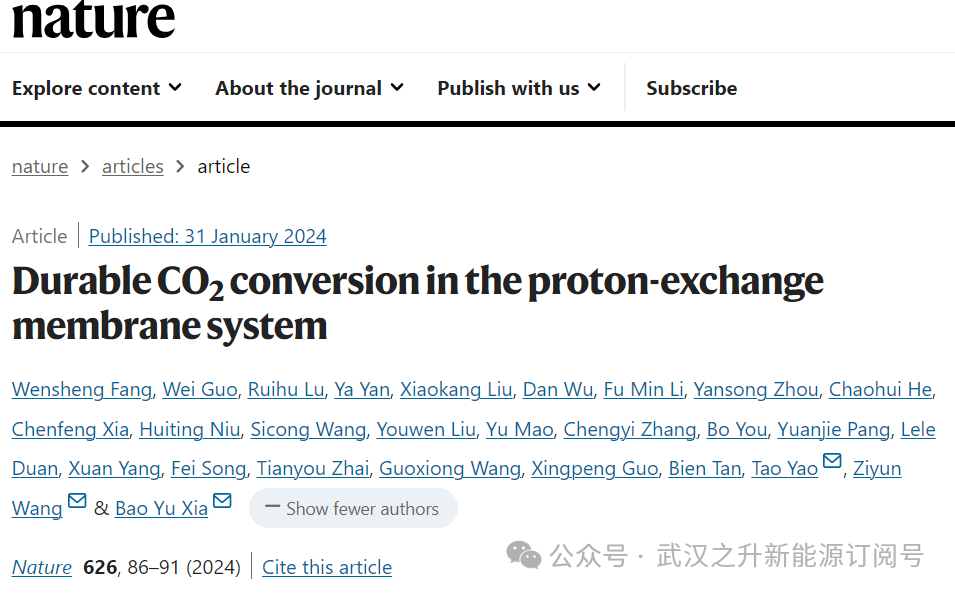
碳酸盐沉淀的问题阻碍了高效和可扩展的二氧化碳转化的发展,如计算的Pourbaix图(图1a)所示,碳酸盐形成发生在很宽的pH范围内。因此,为了避免甲酸生产过程中碳酸盐的形成,需利用先进的电解技术,最终解决碳酸盐沉淀问题,在质子交换膜(PEM)系统下的强酸中进行二氧化碳还原反应(CO2RR)被认为是潜在的解决方案之一。作者开发了一种用于酸性CO2电解的PEM电解槽,其中氢气氧化反应(HOR)发生在阳极,CO2在阴极直接转化为甲酸(图1b)。阴极回收Pb (r-Pb)催化剂是从铅酸电池废料中获得的,可以在工业上以公斤级甚至吨级制备(图1c,插图)。该r-Pb催化剂是铅和硫酸铅(Pb-PbSO4)的复合物,其x射线衍射(XRD)如图1c所示。通过调整球磨时间,可以控制r-Pb的粒径从微米到纳米(图1d和图2)。低温电镜观察显示两个晶格条纹,分别为0.21 nm和0.29 nm,归属于PbSO4和Pb(图1e)。
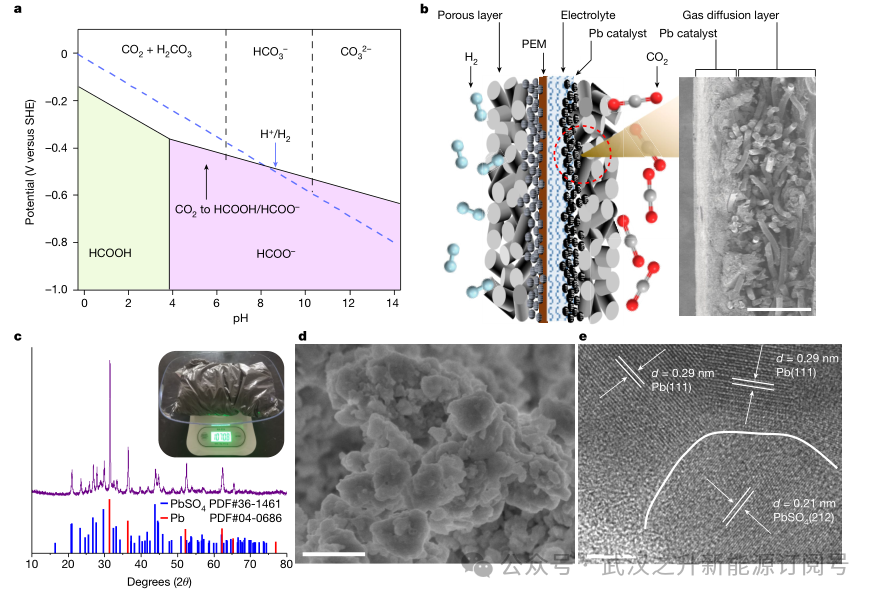
图1. (a) CO2RR中甲酸和氢气生成的Pourbaix图 (H2CO3 KΘa1 = 4.2 × 10−7;KΘa2 = 4.7 × 10−11;SHE,标准氢电极;KΘa1和KΘa2分别为碳酸的第一和第二解离平衡常数)。(b) 用于CO2RR的PEM电解槽原理图。右图为制造的阴极电极的SEM横截面图像。r-Pb催化剂的XRD图 (c)、SEM (d)和TEM (e)。

图2. 不同球磨时间后r-Pb的SEM图像。(a) 0 min, (b) 10 min, (c) 20 min, (d) 30 min, (e) 40 min, (f) 50 min。
【DFT计算】
使用Vienna Ab – initio Simulation Package对周期板模型进行了自旋极化DFT计算。在Perdew-Burke-Ernzerhof泛函中的广义梯度近似中描述了电子交换和相关相互作用,并使用投影增广波方法计算了电子–离子相互作用,其平面波基集由450 eV的动能截止值定义。采用Grimme等人开发的DFT-D3方法处理吸附剂与表面之间的远距离色散相互作用。利用Monkhorst-Pack方案中3 × 3 × 1网格的k点采样进行优化。当电子自一致迭代和力分别达到10−5 eV和0.02 eV Å−1时,完成几何优化和能量计算。Bader电荷分析用于研究所有系统中原子的电荷状态。
自由能变化的计算公式为ΔG = ΔE + ΔEZPE – TΔS,其中ΔE为DFT计算得到的能量变化,ΔEZPE为吸附态与气相之差,ΔS为吸附态与气相的熵差。基于谐波近似和固定催化剂板,利用中间物质的振动频率计算零点能量和熵。此外,根据计算氢电极方法,质子–电子对的能量等于氢分子能量的一半。
为了模拟PbCO3和Pb的表面,首先测量了不同晶格面的表面能。表面能越低,相应的晶格面越稳定。选择稳定的PbCO3(010)和Pb(111)分别代表PbCO3和Pb金属的表面。Pb(111)表面为4层p(2 × 2)超级单体,上层3层松弛,下层1层固定。对于PbCO3(010)表面,模拟了一个两层c(1 × 2)超级单体,其中上层有3个松弛层,下层有0.5个固定层。为了避免周期性相互作用,在z方向上设置了15 Å真空层。
【电极制备】
所使用的所有催化剂都是环保的,来自于废旧铅酸电池,这些电池要么来自于天然废弃的电动自行车,要么来自于LAND测试站模拟的充电和放电电流为30 mA,充放电截止电压为4.8和4.0 V的电池。然后,通过拆卸废铅酸电池并用去离子水清洗三次来获得铅板。然后在真空烘箱中干燥10小时,并进行从板上去除网格的下一个程序。将上述材料在砂浆中手工研磨10 min,得到预循环Pb催化剂。最后将预循环Pb在球磨机中进行0、10、20、30、40和50 min的球磨,得到r-Pb。
【主要内容】
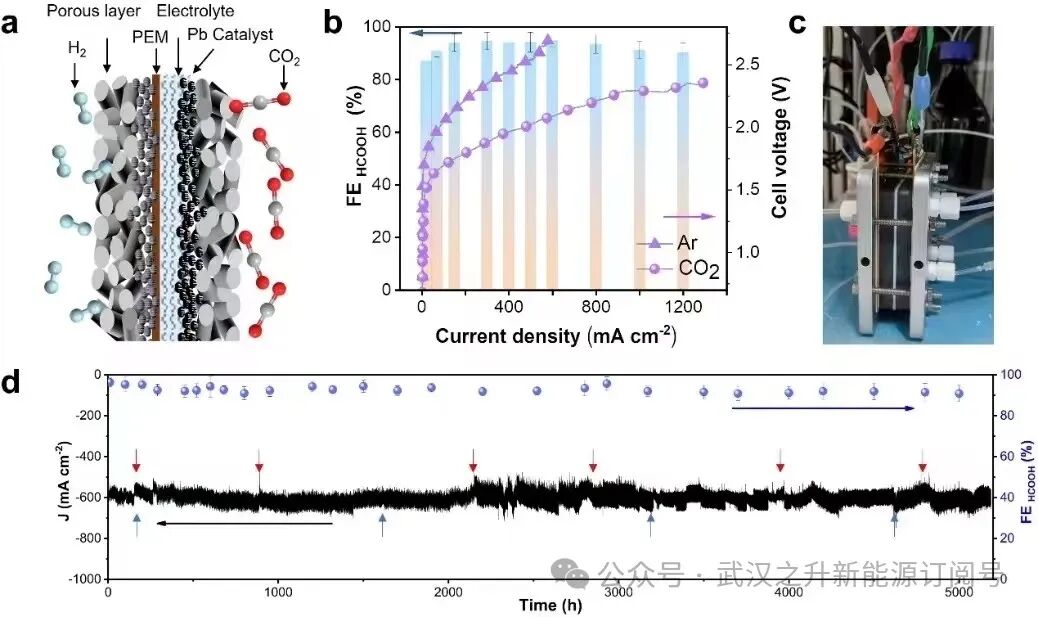
为了证明了PEM系统的可行性与可扩展性,作者在PEM电解槽(pH 1.0)和阳极HOR中评估了阴极CO2RR在r-Pb催化剂上的电化学活性。如图3a所示,通过调整研磨时间生产的优化电极,在2.4 V电池电压下,甲酸法拉第效率超过93%,电流密度高达1.2 A cm−2。r-Pb电极表现出优异的pH耐受性,在pH值为0时具有64%的法拉第效率(由于HER动力学过度),在所有其他pH值下具有91%以上的法拉第效率(图3b)。然而,在碱性电解质(pH 14.0)中,碳酸盐沉淀问题导致50%的显著碳损失,而酸性PEM系统的碳损失非常低,不到1%(图3b)。此外,在该PEM装置中,通过将CO2气体流量优化至3标准立方厘米min-1 (sccm),实现了约91%的CO2单次转换效率(图3c)。更重要的是,该器件可以在2.2 V电压和600 mA cm-2电流密度下稳定工作5200小时(图3d)。PEM系统表现出的卓越稳定性主要归功于化学稳定的r-Pb催化剂,该催化剂在铅酸电池中可以存活数万小时。这种r-Pb催化剂即使在300天后也表现出几乎未衰减的化学稳定性(图3e),并且与包括启动/关闭在内的实际操作具有出色的兼容性(图3e,插图)。此外,三相界面电润湿引起的电解液溢出往往会导致系统失效。在这里,通过每隔200 h使用聚四氟乙烯(PTFE)乳液对气体扩散电极进行再处理来保持三相界面的稳定性(图3f)。此外,作者还通过PTFE膜负载的r-Pb催化剂实现了2000 h的稳定性,无需任何后处理操作。阳极反应在整个系统中起着至关重要的作用,影响电池电压、反应速率、产物分布、膜稳定性和系统寿命。通过利用HOR而不是在阳极进行水氧化反应(WOR),能够降低总电压,更重要的是能够避免产生有害的过氧化氢,过氧化氢会降解甚至破坏PEM。这最终促成了膜的耐用性和PEM系统的长使用寿命(图3g)。因此,面积为5 × 5 cm2的PEM反应器可以在大约2.7 V的电压下实现15 A的电流,生成甲酸的法拉第效率超过91%,证明了PEM系统的可扩展性。
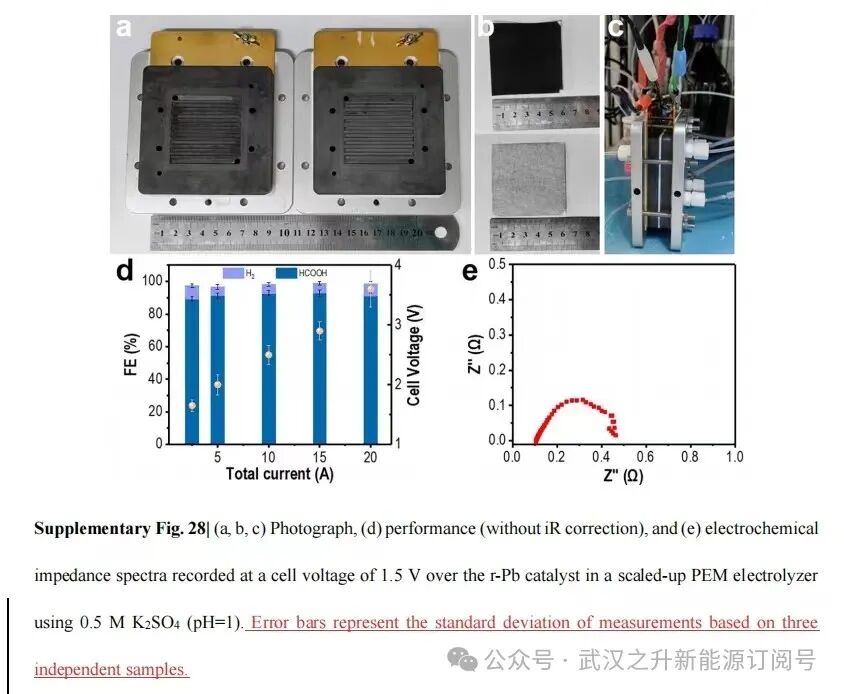

本文所用CO2RR反应器(膜电极液流反应器)由武汉之升能源有限公司提供
图3. (a) 在酸性PEM电解槽中,CO2和Ar气氛下r-Pb的线性扫描伏安(LSV)曲线,以及不同电流密度下CO2RR对r-Pb甲酸的法拉第效率(FE)。(b) 变pH下电流密度为600 mA cm−2时甲酸的法拉第效率和相应的碳损失率。(c) 变CO2流量下r-Pb催化剂的单道转化效率(SPCE)。(d) 电池电压为2.2 V时PEM反应器的电化学稳定性。红色和蓝色箭头所示位置分别代表电解液和反应气体的替换位置。(e) 在PEM反应器中记录的600 mA cm−2下r-Pb催化剂的化学稳定性。其中,稳定性测试由启动/关闭实验组成。(f) 2.2 V下CO2电解2000 h前后气体扩散电极的接触角(CA)测量。(g) 在2.2 V电压下,PEM系统中电解500小时后的原子力显微镜图像。误差条表示基于三个独立样本的测量的标准差。图3g中的Ra表示平均粗糙度。
为了研究r-Pb催化剂的动力学行为,探究高效的CO2电解催化剂中结构–功能关系的重要影响。作者进行了一系列的原位表征,在CO2电解过程中,r-Pb电极的结构由Pb – PbSO4转变为Pb – PbCO3(图4a)。扫描电镜(SEM)图像显示了PbCO3的片状堆积结构的形成和元素分布(图4b和图5a-c)。透射电子显微镜(TEM)图像显示了催化剂的内部结构,表现为Pb和PbCO3的混合物(图5d-f)。原位XRD和拉曼表征证实,结构转变与反应过程有关,而与外加电压无关(图4c、d)。在二氧化碳电解1800秒期间,PbSO4在977 cm−1处的主要拉曼峰逐渐减弱并消失,同时在1055 cm−1处出现碳酸盐的峰值(图4d)。然后,软x射线吸收光谱显示,氧的状态随着CO2RR发生了明显的变化(图6),在短时间内从一条平坦的线到一个明显的峰,然后稳定在534 eV,归属于PbCO3。在PbL3边缘进行了原位扩展x射线吸收精细结构(EXAFS)测量,以研究工作条件下Pb的化学状态。x射线吸收近边结构(XANES)谱图显示Pb的价态逐渐降低(图4e)。随着外加电位从−1.3 V增加到−1.7 V,吸收边出现低能移,表明在工作条件下Pb的价态逐渐降低。同时,所有吸收边的位置都高于Pb箔,表明Pb的价态保持在零以上,EXAFS进一步证实了这一点。如图4f所示,在不同施加电位下获得的所有光谱中都可以看到四个主导峰。位于1.5和2.2 Å的两个峰对应于PbO、PbCO3和PbSO4中的Pb – O/C,而位于2.7和3.4 Å的两个峰对应于金属Pb中的Pb – Pb。随着外加电位从−1.3 V增加到−1.7 V, Pb- O/C峰减少,Pb- Pb峰增加,表明金属Pb的百分比增加。此外,在2.2 Å附近的特征峰位置表现出较高的红移趋势,结合之前的电化学数据结果表明,催化剂发生从PbSO4到PbCO3的结构变化。综上所述,原位XAFS表征证实了r-Pb电极上Pb(II)和金属Pb共存并逐渐演化,进一步说明了CO2电解过程中Pb – PbSO4向Pb – PbCO3复合材料的动态结构转变。其他催化剂也观察到类似的结构转变,表明Pb-PbCO3复合材料的催化结构在PEM体系中是热力学稳定的。这些发现对r-Pb催化剂的动力学行为提供了重要的见解,强调了在设计高效的CO2电解催化剂时理解结构–功能关系的重要性。
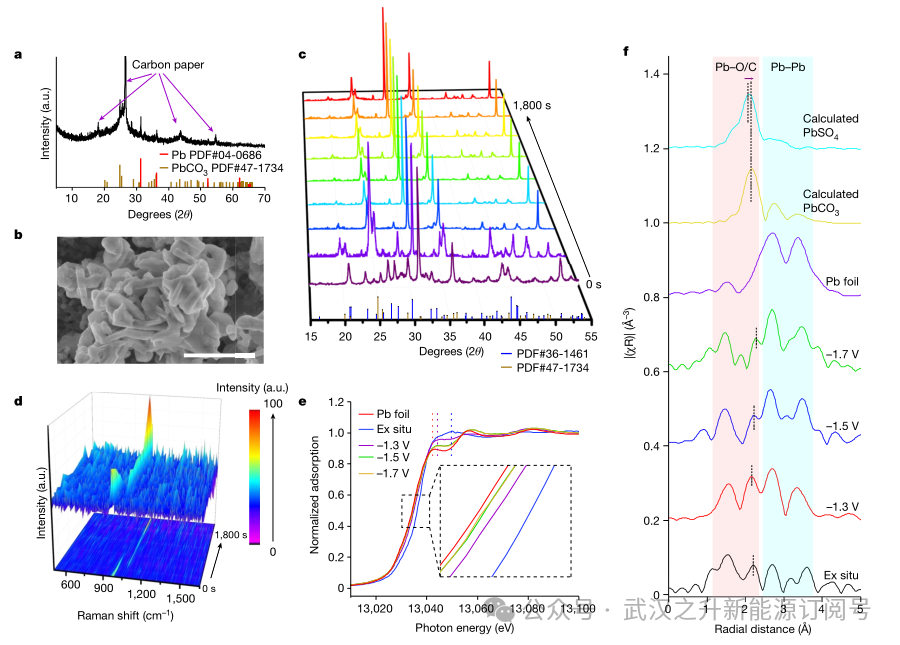
图4. (a, b) CO2还原100 h后r-Pb催化剂的XRD谱图(a)和SEM图(b),(c, d) r-Pb催化剂在– 1.7 V与RHE下的原位XRD谱图(c)和拉曼光谱(d)。(e, f) 在不同施加电位(相对于RHE)下r-Pb催化剂上PbL3边缘记录的原位XANES光谱(e)和相应的傅立叶变换的k2加权EXAFS光谱,以及计算得到的PbSO4和PbCO3光谱作为参考(f)。

图5. (a-b) CO2RR 100 h后r-Pb的SEM图,(c) EDS图,(d-f) TEM图。
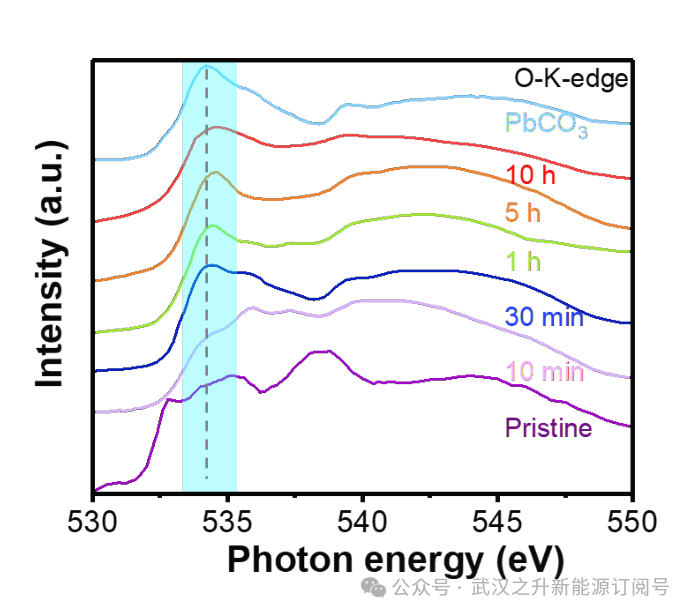
图6. 在2.2 V电池电压下,不同CO2RR次数后r–Pb的Ex原位O – K边近边x射线吸收精细结构(NEXAFS)光谱。534 eV处的峰属于碳酸盐的特征峰。
作者又通过密度泛函理论(DFT)计算来研究CO2RR在r-Pb电极上的高活性和稳定性,特别是关注Pb-PbCO3界面。最初,作者假设废弃铅酸电池中的r-Pb在PEM系统中发生固相转变(图8a),并确定与Pb相关的主要物质为金属Pb、PbCO3和Pb – PbCO3的界面。因此,CO2RR过程在具有潜在局部稳定环境的Pb位点上进行了研究(图7),包括金属Pb(111)、PbCO3(010)、PbCO3 – VCO3和Pb – PbCO3界面。发现,CO2RR首先是化学吸附,然后是电还原;分别用O-C-O角和Pb氧化态来描述化学和电化学过程的程度。由于Pb-PbCO3具有较强的CO2吸附能力和碳酸盐构型的化学活性,其CO2RR向HCOOH的自由能上移最低,表明其CO2RR理论活性较高(图8b)。同时,Pb-PbCO3对*CO2的吸附能为– 0.62 eV,有利于CO2的吸附而不利于*H的吸附。此外,在接下来的两个加氢步骤中,能量的增加幅度分别为0.31和0.48 eV,低于HER (0.50 eV)。这进一步确保了对CO2RR的优越催化性能。即使电位达到– 1.0 V(图8c),与*H吸附相比,Pb-PbCO3位点仍然优先考虑与电位无关的*CO2吸附。电化学CO2RR步骤相对于HER保持较低的能量变化,表明与HER相比,CO2RR是主导反应,使CO2RR能够在酸性电解质中运行,而无需考虑碳酸盐。
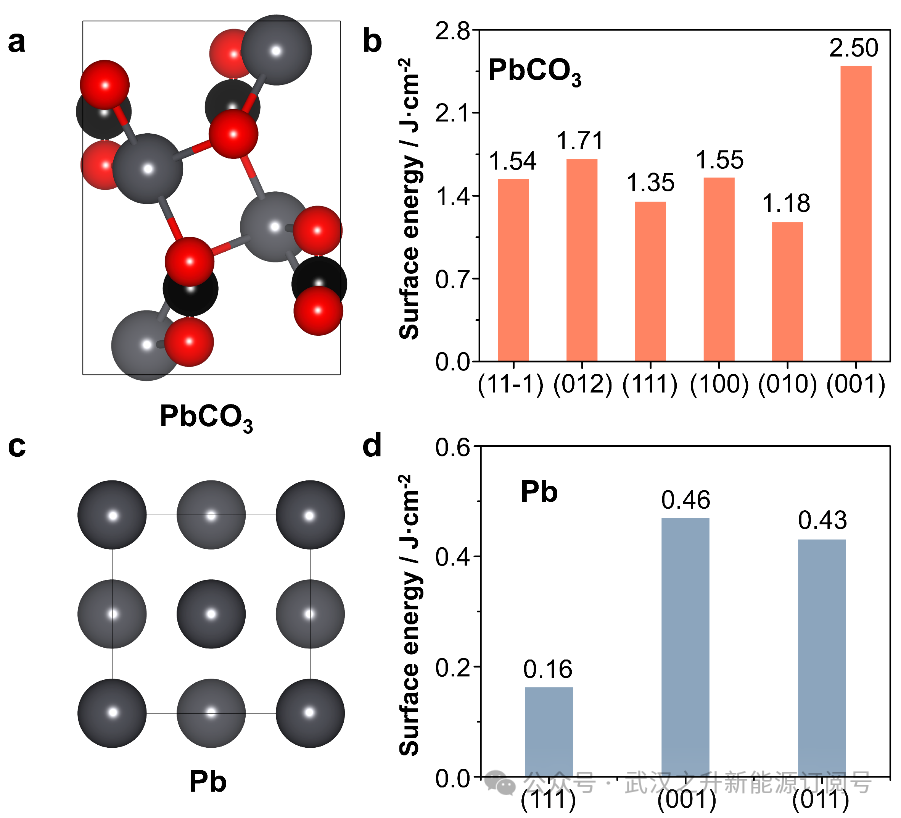
图7. (a) PbCO3晶格的侧视图。黑色:C;红色:O;灰色:Pb。(b) PbCO3中各晶格面的表面能。(c) Pb晶格的侧视图。灰色:Pb。(d) Pb金属中各晶格面的表面能。
作者研究了r-Pb相变过程中Pb-PbCO3的位点演变(图8d)。PbHCO3质子耦合电子转移的形成能为1.1 eV,在相变过程中形成能最高。在– 1.2 V的工作电位下,从热力学的角度来看,这一步骤可以自发发生(图8e)。随后,第二次加氢释放HCOOH形成PbO(*)构型。值得注意的是,与PbO(*)上的第三次加氢相比,CO2分子与PbO(*)结合更强,因此CO2分子在PbO上以碳酸盐形式活化。其次,向HCOOH的双电子CO2RR倾向于发生在PbO(*)位点上,而不是随后PbCO3向金属Pb的还原。这一发现表明,CO2RR可以避免PbCO3的还原,保持稳定的Pb-PbCO3界面。同时,金属Pb可以随着CO2和电解液的流动自发转化为PbCO3。Pb-PbCO3相变循环(图8a)动态生成了Pb-PbCO3的活性位点,理论上保证了r-Pb催化剂对CO2RR的高稳定性。
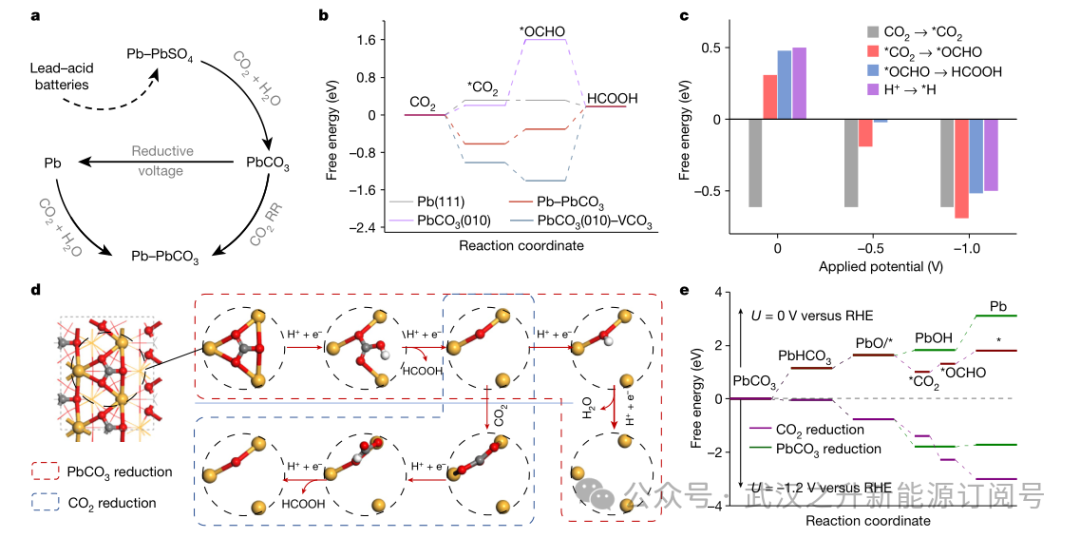
图8. (a) r-Pb催化剂的相变示意图。(b) 活性Pb原子沿CO2RR的自由能谱,PbCO3(010)-VCO3表示PbCO3(010)中碳酸盐的空位。(c) 不同作用势下反应的自由能变化。(d) 固体转化诱导催化机理示意图。白色、灰色、红色和橙色球体分别代表H、C、O和Pb原子。(e) 相变过程中PbCO3还原和CO2还原的自由能谱。
【主要结论】
在这项工作中,作者设计、开发并测试一种质子交换膜系统。该系统通过源自废铅蓄电池的催化剂将CO2还原为甲酸。首先证明了PEM系统的可行性与可扩展性,通过在PEM电解槽(pH 1.0)和阳极HOR中评估了阴极CO2RR在r-Pb催化剂上的电化学活性,面积为5 × 5 cm2的PEM反应器可以在大约2.7 V的电压下实现15 A的电流,生成甲酸的法拉第效率超过91%。然后,作者通过一系列原位表征探究CO2RR过程催化剂中的结构–功能关系,并通过密度泛函理论(DFT)计算来研究CO2RR在r-Pb电极上的高活性和稳定性,揭示了新的动态固相诱导的晶格碳活化机制促进了该过程的二氧化碳高效转化,Pb-PbCO3相变循环动态生成了Pb-PbCO3的活性位点,理论上保证了r-Pb催化剂对CO2RR的高稳定性。通过关键催化材料开发、膜电极系统设计,以及电极反应协同优化等策略,构筑了新型二氧化碳电解系统,实现了二氧化碳电解系统高效长寿命服役。当CO2还原与氢氧化耦合时,甲酸的法拉第效率超过93%。该系统与启动/关闭过程兼容,在电流密度为600 mA cm -2和电池电压为2.2 V的情况下,二氧化碳的单通转换效率接近91%,连续工作时间超过5200小时。这些结果表明PEM系统能够有效和稳定地将二氧化碳转化为甲酸,这种卓越的性能将有助于推动碳中和技术的发展。
【文章引用】
Fang, W., Guo, W., Lu, R. et al. Durable CO2 conversion in the proton-exchange membrane system. Nature 626, 86–91 (2024).