
第一作者:Ouwen Peng
通讯单位:新加坡国立大学和南方科技大学
通讯作者:Chun Cheng,Zhongxin Chen,Kian Ping Loh
【成果简介】
水相中电化学氨合成的一个常见挑战是竞争析氢反应(HER)消耗法拉第电荷,这降低了所需转化的法拉第效率,即硝酸盐还原反应(NO3RR)转化为铵。当单相催化剂在高电流极限下运行时,这个问题特别严重,因此需要协同工作用于氢获取和脱氧的助催化剂系统来促进NO3RR而不是HER。在此,作者选择了一种众所周知的HER催化剂Mo2C,并研究了金属掺杂如何将其动力学从HER主导的途径转换为NO3RR主导的途径。在Mo2C的3.8wt%Ru掺杂下,在16cm2的流动电解槽中实现了75%的硝酸盐(0.1M)到铵的单程转化,对应于在2V的全电池电压下9.07mmol h−1的铵产率。DFT计算和动力学同位素实验证实,基质中的钌掺杂剂在NO3RR过程中充当吸附氢的汇点,以促进*NO3和*NO2在Ru−Mo共催化位点上的协同脱氧。作者的研究表明,优化共催化系统中的氢气获取和脱氧反应是电化学合成的有效策略。
【研究背景】
从含氮原料(如N2、亚硝酸盐和硝酸盐)中电化学生产氨是Haber−Bosch工艺的一种新兴替代方法,因为其操作条件温和且与可再生能源兼容。从热力学和经济角度来看,硝酸盐还原反应(NO3RR)的活化能比N2气体低得多(204比941 kJ mol−1),因此很有吸引力。工业污水和生活污水中硝酸盐的丰度(高达2M)为回收利用提供了无限的库存,可作为有用的原料。NO3RR催化剂可用于关闭全球氮循环,实现更可持续的未来。然而,其缓慢的八电子介导的还原途径在选择性生产铵以对抗其他副产物(如亚硝酸盐、NO、N2、N2O和肼)方面带来了重大挑战。析氢反应(HER)和NO3RR之间的竞争也会导致在更负的电压下性能下降,从而阻止反应物的高转化率。从根本上讲,NO3RR分为从硝酸盐到*NO(或*N)的一系列脱氧步骤,以及随后的氢化步骤以产生氨。在每个基本步骤中,氢在氢供体(*H)和受体(吸附质)之间发生转移。因此,NO3RR的反应动力学与氢转移的性质和可用氢供体的密度密切相关。为了合理的催化剂设计,需要深入了解NO3RR中的基本步骤,例如,理想的NO3RR催化剂应优选在表面共活化*H和*NO3,同时避免*H快速重组为H2或在高硝酸盐浓度下被中间体中毒。
吸附氢的利用在脱氢、氢化和氢解等广泛的反应中是普遍的,这些反应通常由Pt族金属(Ru、Rh、Pd、Os、Ir和Pt)催化。然而,水溶液中Pt类金属上*H的高覆盖率在动力学上有利于HER而不是NO3RR。受Levy和TMC中母体金属由于添加C原子而增加的d-电子密度的启发。先前对TMC基电催化剂的研究主要集中在HER、氧还原和CO2还原反应尚未扩展到NO3RR。一个主要原因在于TMC上硝酸盐的活化较差。TMCs较差的固有能力提供了一个机会来研究金属掺杂或改性将如何改变吸附的氢的转移动力学,从而使反应动力学可以在低施加电压下“切换”为氨的电合成。
在这里,作者选择了一种典型的类Pt电催化剂,β相碳化钼(Mo2C),以研究单原子助催化剂的掺杂如何改变反应动力学并促进NO3RR。众所周知,纯Mo2C的动力学以强HER为主;因此,它不是NO3RR或其他竞争反应的选择。为了将动力学从HER转变为NO3RR,作者系统地研究了Ru在β相Mo2C基体中的掺杂效应。结合密度泛函理论(DFT)研究和动力学同位素分析,发现Mo2C基体中的Ru掺杂剂有助于NO3RR脱氧步骤而不是HER的Heyrovsky步骤中吸附氢的获取和转移。在3.8wt%的Ru掺杂下,Ru掺杂的Mo2C在组装的16cm2流动电解槽中,在2V的全电池电压下,表现出创纪录的75%的硝酸盐到铵的单程转化率,以及90%的法拉第效率(FE)和9.07mmol h−1的铵产率。
【主要内容】
Ru1/Mo2C催化剂的表征
通过改进的溶胶–凝胶法在石墨毡载体上制备了一系列Ru负载量为0.2−3.8 wt%的Ru掺杂Mo2C催化剂(Ru1/Mo2C)。图S1中的扫描电子显微镜(SEM)图像显示,石墨毡上的每根碳纤维上都有均匀的催化剂涂层,使其具有在流动电解槽中使用的抗渗性。如图1A和S3所示,在高角度环形暗场扫描透射电子显微镜(HAADF-STEM,用圆圈标记)图像中识别出原子分散的Ru掺杂剂。图1D中的放大图证实了Mo2C晶格中Ru取代Mo,这与图1E、F中模拟的HAADF图像和原子模型非常一致。Ru原子取代Mo归因于在凝胶化过程中羰基Mo溶胶前体掺入Ru3+离子。由于Mo2C初始核形成的强烈趋势,Ru发生了表面富集。空间分辨电子能量损失谱揭示了Ru(2838eV处的L3edge)、Mo(M-edge和L-edge),和Ru1/Mo2C中的C(K-edge)元素(图1B的插图)。这进一步得到了图S2中能量色散X射线光谱(EDS)图谱的均匀分布以及图S4和S5中没有Ru纳米颗粒聚集的高分辨率TEM图像的支持。在选区电子衍射(SAED)中发现了典型的Mo2C的六边形图案,并且没有观察到Ru的结构畸变。
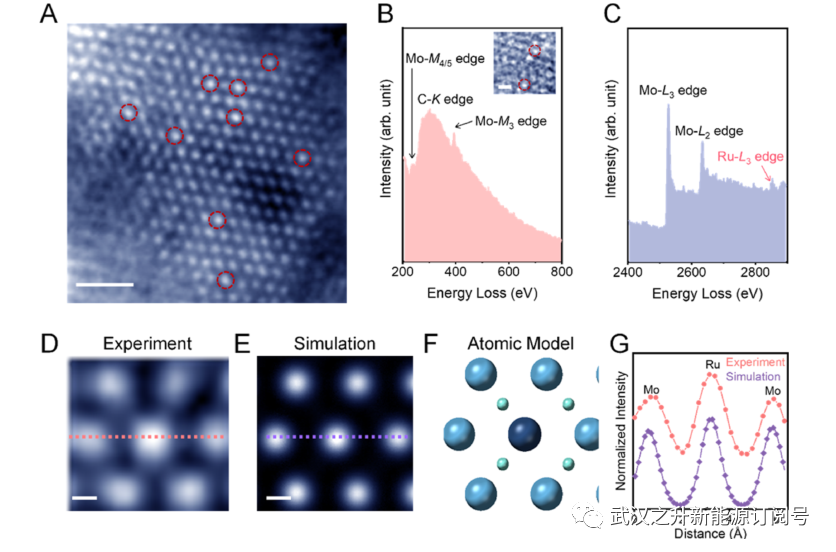
图1. Ru掺杂Mo2C基体的微观证明。(A) Ru1/Mo2C的原子分辨率STEM-HAADF图像。(B,C)Ru1/Mo2C中所选区域的电子能量损失光谱(EELS)(插图)。红色圆圈突出显示了可能的钌掺杂剂。(D)位于Mo柱中的Ru原子的放大视图。(E)相应的HAADF图像模拟和催化剂的(F)原子模型。比例尺:A,1 nm;B、 D,E,0.1nm。(G)沿着实验图像和模拟图像中指示的线的强度分布。
通过扩展X射线吸收精细结构(EXAFS)光谱研究了Ru1/Mo2C的局部键合环境,如图2所示。图2B中原始Mo2C的Mo K边缘光谱与其在六方β相中的键合环境一致。根据表S1中的拟合结果,每个Ru原子在第一配位壳中与三个(cal.2.7)碳原子(Ru-C,2.06Å。)在第一配位壳层中以及在第二壳层中被另外六个(cal.5.8)钼原子(Ru−Mo,3.10Å)包围。这对应于1.47、2.24处的散射路径,图2C中Ru1/Mo2C.的Ru K-边光谱的实验和拟合曲线中的2.61Å。基于EXAFS数据(支持信息中的详细解释),排除了碳层中取代的可能性(第一壳层中2.12Å处有六个Ru−Mo)。Ru1/Mo2C没有观察到金属Ru颗粒的峰,揭示了Ru1作为隔离的掺杂剂存在。Ru催化剂的单原子性质也由图2D中的X射线衍射(XRD)支持,其中在高达3.8wt%的Ru负载量的情况下未检测到本体金属峰。如图S7中的XRD峰和STEM图像所示,当Mo2C中的Ru≥6wt%时,Ru纳米颗粒发生聚集。
利用X射线吸收近边结构(XANES)和X射线光电子能谱(XPS)验证了Ru1/Mo2C中Ru的电子结构和氧化态。如图2A所示,在Ru K-edge XANES光谱中,Ru1/Mo2C催化剂的白线强度远低于RuO2和RuCl3前体,略高于Ru箔,这表明Ru.44的氧化态在0和+2之间。图2E中的Ru 3p核级XPS光谱证明了这一点。在462.8和485.1 eV处观察到可归属于Ruσ+的自旋轨道双峰的两个峰,其结合能在Ru(IV)和Ru(0)之间。类似地,接近C1s信号的Ru 3d核能级光谱显示出Ru 3d5/2的结合能为280.6eV。这些结果证实了由于Ru掺杂剂和Mo2C载体之间的强电子相互作用而产生的部分正电荷(Ruδ+)。Ru的表面富集也通过XPS得到证实,其中Ru与Mo的比率远高于本体中的理论值(表S2和S3)。同时,与图S8中的Mo 3d XPS光谱和Mo K-edge XANES光谱相比,Mo的氧化态几乎保持不变,表明低浓度的Ru掺杂剂不会影响Mo2C的化学环境。
H电池中的电化学硝酸盐还原
NO3RR实验在环境条件下在氩中的定制H型电池中进行。直接使用Ru1/Mo2C涂层石墨毡(3 mg cm−2,~0.11 mg Ru cm−2)作为工作电极。为了进行比较,将20 wt%的Ru/C催化剂以0.5 mg cm−2的质量负载(即相同的Ru负载)滴涂到石墨毡上。
首先在含有1M KOH和各种浓度的KNO3的电解质中进行线性扫描伏安法(LSV),以验证对NO3RR的催化活性。如图S9所示,在存在或不存在1M KNO3的情况下,原始Mo2C的LSV曲线几乎没有差异,这表明HER的干扰很强。这显著降低了铵转化的FE,并排除了高反应转化率,尤其是在更负的电压下,由于静电相互作用,带正电的质子的还原比带负电的硝酸盐更有利,并且在硝酸盐的稀释浓度下,HER的影响不容忽视。形成鲜明对比的是,与HER相比,在3.8wt%的Ru1/Mo2C催化剂上更容易发生硝酸盐还原,其中在添加硝酸盐时观察到显著增强的电流密度。在1M KNO3中,起始电位正移超过300mV。最重要的是,从起始电位和电流密度来看,Ru1/Mo2C的NO3RR性能远优于商用的20wt%Ru/C。
然后通过计时电流法(CA)在1M KOH和0.1M KNO3中从0.1到−0.5V相对于RHE的测量来验证铵的生产率。使用公认的吲哚酚蓝比色法来量化电解质中产生的铵,以测定FE、部分电流密度和产率(详细信息见图S13和支持信息的实验部分)。如图S14所示,相对于RHE,3.8wt%的Ru1/Mo2C在0.1至-0.2V的电势范围内对铵表现出超过80%的高FE值;在图3A中,它在−0.2 V下达到317.0 mA cm−2的最大分电流密度和1.48 mmol h−1 cm−2(或25.1 mg h−1cm−2)的铵产率,这优于Mo2C、0.7 wt%Ru1/Mo2C和20 wt%Ru/C。值得注意的是,结果表明,优化的Ru负载量为3.8wt%,尽管低浓度的Ru掺杂仍然会提高NO3RR的反应性。当在催化剂制备过程中引入过量负载(超过6wt%)的Ru时,Ru纳米颗粒在Mo2C中更负并且可以观察到可见气泡时,就会发生析氢。这导致FE和铵的产率降低。尽管如此,如表S5所示,3.8wt%Ru1/Mo2C的性能仍然优于其他已报道的催化剂。
为了证实NO3RR性能的可靠性,作者还通过1H核磁共振(NMR)光谱定量了铵的量。54所有结果与吲哚酚蓝法的结果一致(图S13)。此外,图3B中使用K15NO3作为反应物的同位素标记实验证实,铵的来源是硝酸盐,而不是电解质和空气中的杂质。在−0.2 V下电还原30分钟后,使用K15NO3在1H-NMR中可以看到15NH4+的典型双态,而使用传统KNO3的对照实验将产生14NH4+的三态。定量同位素测量的假阳性测试支持了这一点;还进行了空白研究,以排除杂质的影响(图S16)。
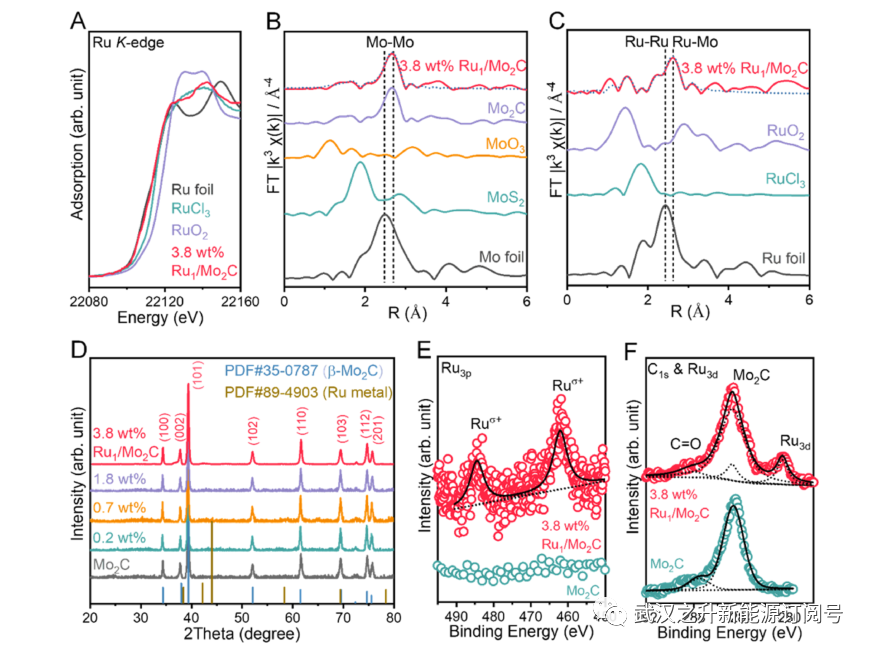
图2. Ru1/Mo2C催化剂的表征。(A) Ru K边XANES光谱。(B,C)各种催化剂的FT-EXAFS光谱。虚线表示FT-EXAFS光谱的拟合结果。(D) Ru1/Mo2C催化剂在不同负载下的XRD图谱。(E) Mo2C和Ru1/Mo2C的高分辨率XPS Ru 3p光谱和(F)C1s和Ru 3d光谱。
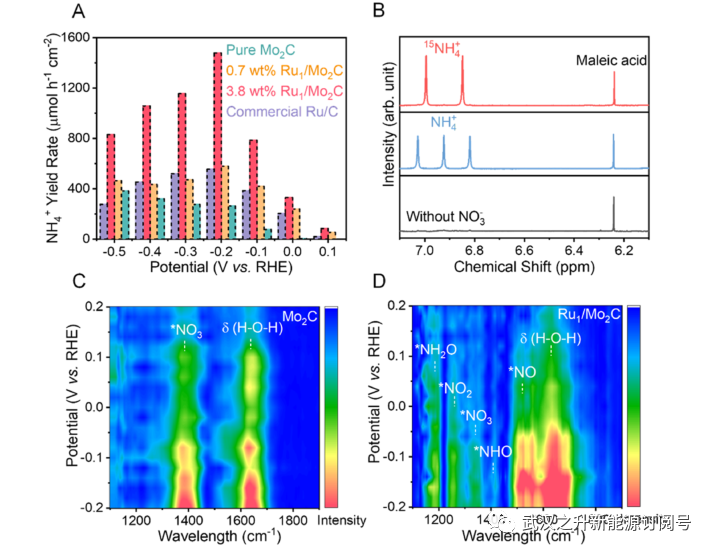
图3. 验证氢电池设置中的NO3RR性能。(A) 各种催化剂在每种给定潜力下的铵生产速率。(B)15N同位素标记实验证明了氮源的来源。反应后使用含15NO3−或14NO3−的电解质收集1H NMR光谱。(C,D)使用Mo2C和Ru1/Mo2C催化剂的NO3RR的原位FTIR光谱。
深入了解NO3RR途径
在纯Mo2C和Ru1/Mo2C催化剂相同的条件下,通过原位傅立叶变换红外光谱(FTIR)探测NO3RR中间体的存在,以研究Ru掺杂剂促进的反应途径的任何变化。如图3C所示,纯Mo2C仅表现出与*NO3吸附质相关的两个峰以及水的弯曲振动。相反,在图3D和S17中的Ru1/Mo2C的光谱中观察到一系列NO3RR中间体,包括1186 cm−1(−N−O−振动)处的*NH2O物种、1248 cm−1处的*NO2、1412 cm−1下的*NHO和1521 cm−1上的*NO。与纯Mo2C相比,*NO3吸附质的强度显著降低,这表明即使在低过电位值下,Ru1/Mo2C表面上的硝酸盐也会快速消耗。因此,该结果表明了一种可能的途径,涉及*NO中间体的产生和随后NO*的氮的氢化,以提供*NH2O和最终的铵。
进行DFT计算以深入了解可能的NO3RR途径,如图4和5A所示。模拟Ru1/Mo2C模型的结构基于图4和5A中表面Ru取代Mo。在表S6中的各种可能构型中,根据DFT,亚Mo的结构被确定为热力学最稳定的。经计算,整个加氢过程(N−O→H−N−O→H2−N−O→N−H3)对Mo2C和Ru1/Mo2C的影响。热力学障碍主要发生在逐步添加吸附的氢和去除。热力学障碍主要发生在脱氧过程中吸附的氢的逐步添加和羟基的去除中工艺(N−O3→HH−N−O3→NN−O2→HH−N−O3→NN−O)。 Ruopants有效地将*NO3的能垒降低为*HNO3(0.83−0.23 eV)和*NO2到*HNO2(1.13−0.52 eV)。DFT研究表明,*H和*NO3在单个活性位点上的共吸附在热力学上是不稳定的。作者假设Ru掺杂剂作为吸附氢的汇点,并提高了脱氧过程中的氢化。
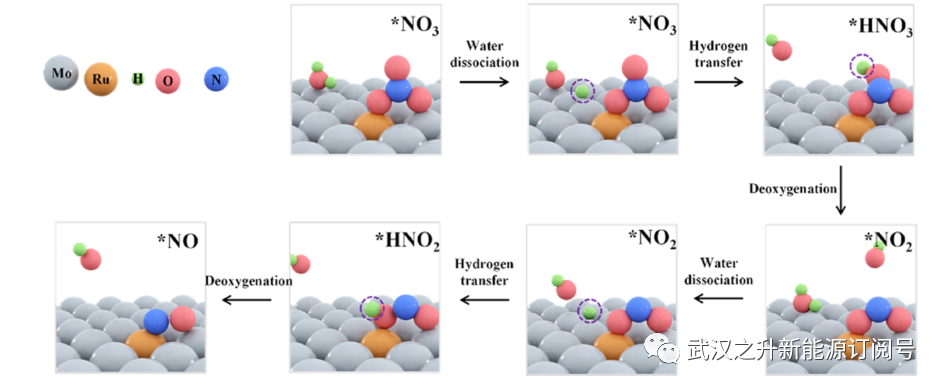
图4. NO3RR的示意图。初始步骤包括水在Mo2C基质上离解以产生活性氢,活性氢被转移到Ru位点用于随后的NO3RR脱氧步骤。配色方案:Mo原子为灰色,Ru原子为黄色,H原子为绿色,O原子为红色,N原子为蓝色。
使用氧化氘进行的动力学同位素实验进一步揭示了Ru掺杂剂促进的氢转移。如图5C、D和S18中的LSV测量所示,HER和NO3RR中Mo2C和Ru1/Mo2C的动力学同位素效应(KIE)值分别在−0.22至−0.38和−0.12至−0.28 V与RHE的范围内进行了分析。所获得的KIE值在所有情况下都大于1,这意味着在速率决定步骤中涉及原子–质子转移而不是电子转移。KIE是原子–质子转移动力学的一般描述符,其中在硝酸盐存在或不存在的情况下,相似的KIE值表明纯Mo2C. HER和NO3RR之间没有动力学偏好。这种相似性也表明,碱水离解产生的质子是速率决定步骤。相反,Ru1/Mo2C的KIE值的显著变化表明,吸附的氢优先转移用于硝酸盐加氢,而不是经过重组形成−0.2 V以上的氢(相对于RHE)。在硝酸盐存在和不存在的情况下,Ru1/Mo2C的KIE差异即使在更负的电势范围内也保持稳定在约0.5,这表明反应界面中存在不饱和覆盖。这种偏好解释了在更正的电势下使用Ru1/Mo2C的铵的相对高的FEs,在效率下降之前,由于在-0.2V与RHE之后Mo2C的无Ru局部结构域上的强HER竞争。研究了纯Mo2C和3.8wt%Ru1/Mo2C的反应动力学对硝酸盐浓度的依赖性。硝酸盐浓度定义了参与NO3RR的硝酸盐量,对HER和NO3RR之间的竞争动力学有着深远的影响(图S19和S20)。从0.1到2 M硝酸盐,Ru1/Mo2C下NO3RR的FE在比−0.2 V vs RHE更负的电势下增加,并且铵产率表现出渐进的增加,在1 M KOH和2 M KNO3电解质中,在−0.4 V vs RHE下达到最大1 mmol h−1 cm−2。如图5E,F所示,绘制了0.1至−0.5 V与RHE的铵的部分电流密度的对数(log | jammonium |)与硝酸盐浓度的对数(log[NO3−])的函数关系。然后仔细检查纯Mo2C和3.8wt%Ru1/Mo2C的log|jamonium|与log[NO3−]的关系图,以确定NO3RR的表观反应顺序。纯Mo2C的NO3−反应级数(趋势线的斜率)从-0.2到0.5 V相对于RHE接近于零(~0.13)。这是因为尽管体电解质中有大量的*NO3,但HER率很高。相反,3.8 wt%Ru1/Mo2C的NOx−反应级数在大多数情况下保持相似的值(cal0.5)。这意味着*NO3由于连续消耗而涉及速率确定步骤。在某些情况下,1−2 M的负序可归因于高浓度NO3−和K+对HER的过度抑制,对NO3RR产生负面影响。
纯Mo2C和3.8wt%Ru1/Mo2C的LSV曲线中的Tafel斜率如图S21所示。从Tafel斜率来看,碱性HER在纯Mo2C上的速率决定步骤是水离解(Volmer步骤,74 mV dec−1);然而,在3.8wt%的Ru1/Mo2C。析氢的Heyrovsky步骤占主导地位。这得到了图5C中模拟的HER途径的支持,其中Ru1/Mo2C中的水离解能垒远低于未修饰的Mo2C(-1.25 vs 0.27 eV)。同样,随后将吸附的氢重组为H2显示出Ru1/Mo2C上更陡的攀升(1.09 vs 0.19 eV)。HER较差的动力学表明,更大比例的吸附氢用于Ru1/Mo2C中硝酸盐的脱氧。
在没有HER和有HER的电位范围内,通过不同硝酸盐浓度下的Tafel斜率分析了吸附氢对NO3RR的影响。纯Mo2C的大Tafel斜率值(>800 mV dec−1)表明,在没有吸附氢的帮助下(没有硝酸盐活化),NO3−的动力学缓慢。相反由于在Ru掺杂剂存在的情况下,速率决定的第一电子转移(水离解的一部分),3.8 wt%Ru1/Mo2C的Tafel斜率在无HER状态下保持在120 mV dec−1。在速率决定步骤中,含有*NO3和*H作为反应物的Langmuir−Hinshelwood机制的框架下,对3.8 wt%Ru1/Mo2C上的NO3RR机制进行了分类。因此,Ru掺杂剂充当Mo2C基质中的汇点,用于获得吸附的氢原子,然后在硝酸盐脱氧步骤中转移,从而使Mo2C的HER活性转变为NO3RR活性。
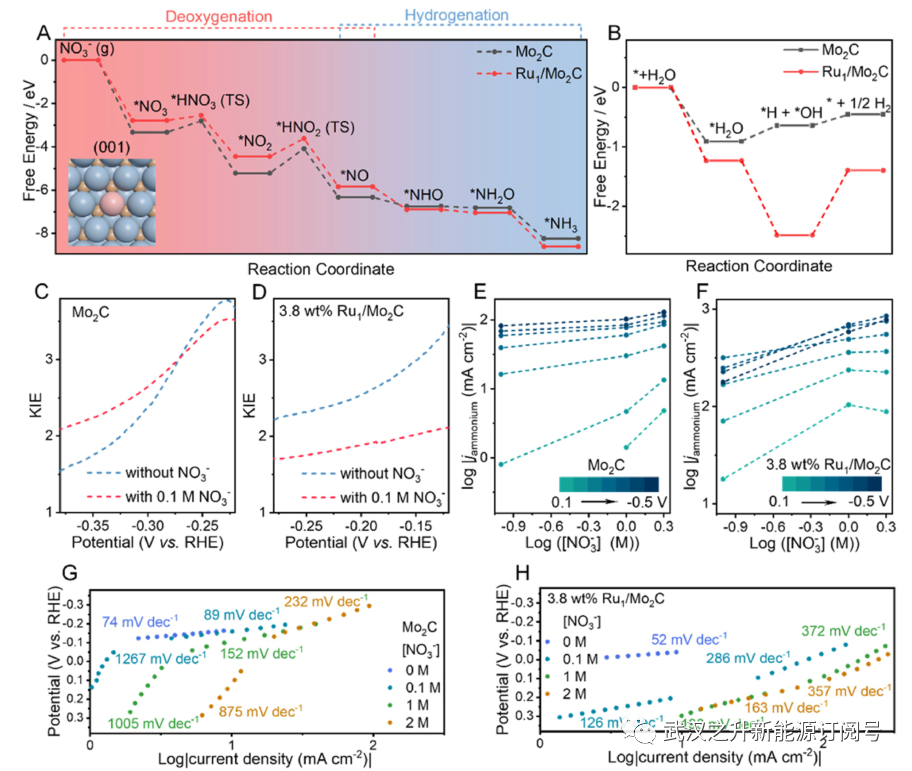
图5. Ru掺杂剂在NO3RR过程中促进脱氧步骤的作用。(A,B)分别在Mo2C和Ru1/Mo2C催化剂上硝酸盐还原和碱性析氢的自由能图。插图显示了用于DFT计算的Ru1/Mo2C的原子结构。配色方案:蓝色的Mo原子,粉红色的Ru原子,棕色的C原子。(C,D)在不存在和存在硝酸盐的情况下Mo2C和Ru1/Mo2C的KIE。(E,F)0.1至−0.5 V的电流密度与RHE的对数,作为硝酸盐浓度对数的函数。(G,H)纯Mo2C和3.8wt%Ru1/Mo2C在1M KOH中与不同浓度(0.1、1和2M)硝酸盐的Tafel图。
16 cm2液流电池中的NO3RR性能
鉴于其优异的NO3RR性能,作者在石墨毡催化剂上使用3.8wt%的Ru1/Mo2C组装了一个16cm2的流动电池,以展示其实际应用的潜力。如图6A,B所示,液流电池在2 mL min−1的1 M KOH和0.1 M KNO3的阴极电解液中表现出优异的性能,在2 V的全池电压下,从硝酸盐到铵的单程转化率高达75%,全池电流为2.13 A(或133 mA cm−2)。这意味着铵的产率为9.07 mmol h−1。流动电解槽在1.5至2.0V的宽电势范围内保持>88%的FE,这意味着Ru掺杂剂对HER的有效抑制。重要的是,流动电解槽的单程转化率可通过阴极电解液的流速高度调节。如图6C和S24所示,在低流速(例如,1 mL min−1)下,传质限制主导了液流电池的性能,其中大部分硝酸盐(90%)转化为铵。HER的发生是由于催化剂表面附近的硝酸盐耗尽,导致FE和铵的产率大大降低。在高流速下,液流电池的反应受到催化剂活性的限制。发现2 mL min−1的流速是平衡传质限制和催化剂反应性的最佳流速。对NO3RR反应器液位的理解对其在废水处理中的实际应用至关重要。最后,如图6D所示,流式电解槽可以提供稳定的性能,在2 V的全电池电压下连续运行25小时后,电流密度和铵的产率下降可以忽略不计。
本文所使用的流动反应池由武汉之升新能源有限公司提供

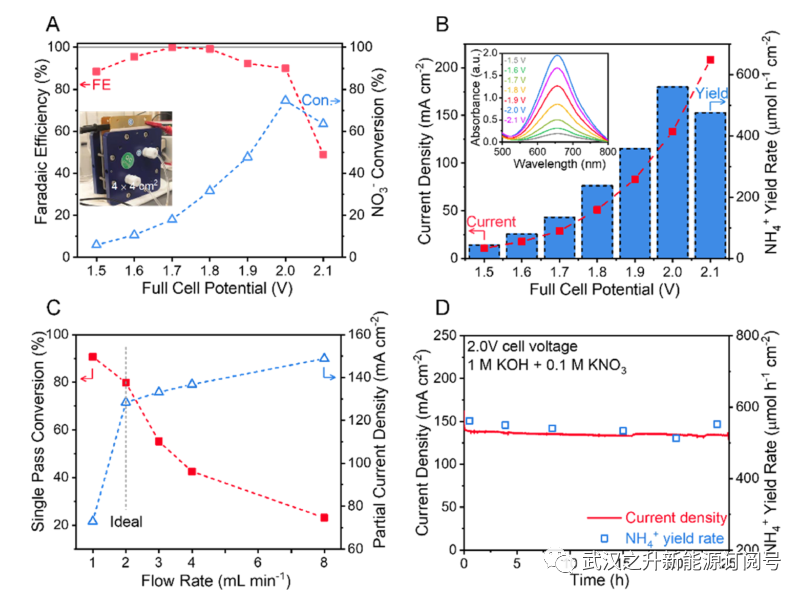
图6. 使用Ru1/Mo2C催化剂进行NO3RR的16cm2液流电池性能。(A) FE和在各种电池电压下的单程转换。插图显示了16cm2液流电池的照片。(B) 在不同电池电压下的总电流密度和铵的生产速率。插图显示了相应的紫外–可见光谱,以确定铵的产率。(C) 硝酸盐的单程转化率和铵的部分电流密度对阴极电解液流速的依赖性。(D) 液流电池的稳定性测试。在一定时期内验证了铵(蓝色开口正方形)的产率。所有实验都是在含有1M KOH和0.1M KNO3的阴极电解液和1M KOH的阳极电解液中进行的。
【主要结论】
作者系统地研究了Ru掺杂剂在β相Mo2C基体中对NO3RR的促进作用。DFT研究和动力学同位素分析证明,Ru掺杂剂比竞争的HER促进了NO3RR中的脱氧步骤。Mo2C基质的高HER活性可以“切换”为具有3.8wt%Ru掺杂剂的高NO3RR活性,在组装的16cm2流动电解槽中,在2V的全电池电压下,实现了创纪录的75%的硝酸盐到铵的单程转化率,以及90%的FE和9.07mmol h−1的铵产率。作者的研究表明,共催化系统有助于最大限度地减少竞争性HER,从而改善电合成反应的FE。助催化剂或工程合金相的空间分布的进一步优化有望提高NO3RR效率。
【文章引用】
Ouwen Peng, Qikun Hu, Xin Zhou, Rongrong Zhang, Yonghua Du, Minzhang Li, Lu Ma, Shibo Xi,Wei Fu, Zong-Xiang Xu, Chun Cheng, Zhongxin Chen, and Kian Ping Loh, Swinging Hydrogen Evolution to Nitrate Reduction Activity in Molybdenum Carbide by Ruthenium Doping,2022,ACS Cataysis.