
通讯作者:王晓龙
通讯单位:华能清洁研究院
DOI:10.1016/j.ensm.2026.105217
工作简介
本综述以失效机制为核心对多硫化物的氧化还原液流电池(PSRFBs)进行分析,将传统的材料聚焦视角转向系统完整性和可恢复性视角。作者团队首先审视长期储能(LDES)不断演变的市场和技术需求,随后系统剖析PSRFBs的四大主要失效模式:界面动力学限制、电解质不稳定性、交叉污染效应以及缺乏有效再平衡策略导致的容量失衡。针对每种失效模式,深入评估了电极、电催化剂、电解质和膜领域的最新进展。本文专门设立章节探讨电解质再平衡策略,并从材料与工程双维度对相应解决方案进行批判性评估。与对称系统(例如全钒液流电池)不同,PSRFBs可能需要更复杂的再平衡策略来应对不可逆的容量衰减。此领域虽尚未得到充分探索,却是实现商业化应用不可或缺的关键方向。本综述最后提出了未来研究方向:应优先考虑工程可扩展性和容量恢复能力,而非短期性能指标,旨在将PSRFBs从一项前景广阔的实验室级技术,转化为未来无碳电网中具有商业可行性的核心基础技术。
研究背景
当前大多数关于多硫化物基氧化还原液流电池的研究主要局限于实验室规模、以材料为核心的探索,例如传统电极改性、催化剂优化、电解质配方设计及隔膜开发。这些研究往往采用碎片化的评估框架,与工程级应用缺乏明确关联。更关键的是,现有文献倾向于孤立地处理每种材料挑战,忽视了在实际系统中这些局限性会协同作用导致系统级故障这一事实,而任何单一材料创新都无法单独解决这些问题。由正负极电解质失衡引发的容量衰减及其相应的再平衡策略仍缺乏充分研究;然而,正是决定PSRFB系统能否实现其理论预测的20000次循环寿命的关键失效模式。当前大多数关于多硫化物基氧化还原液流电池的研究主要局限于实验室规模、以材料为核心的探索,例如传统电极改性、催化剂优化、电解质配方设计及隔膜开发。这些研究往往采用碎片化的评估框架,与工程级应用缺乏明确关联。更关键的是,现有文献倾向于孤立地处理每种材料挑战,忽视了在实际系统中这些局限性会协同作用导致系统级故障这一事实,而任何单一材料创新都无法单独解决这些问题。由正负极电解质失衡引发的容量衰减及其相应的再平衡策略仍缺乏充分研究;然而,正是决定PSRFB系统能否实现其理论预测的20000次循环寿命的关键失效模式。
核心内容
1.水基多硫化物氧化还原液流电池的应用
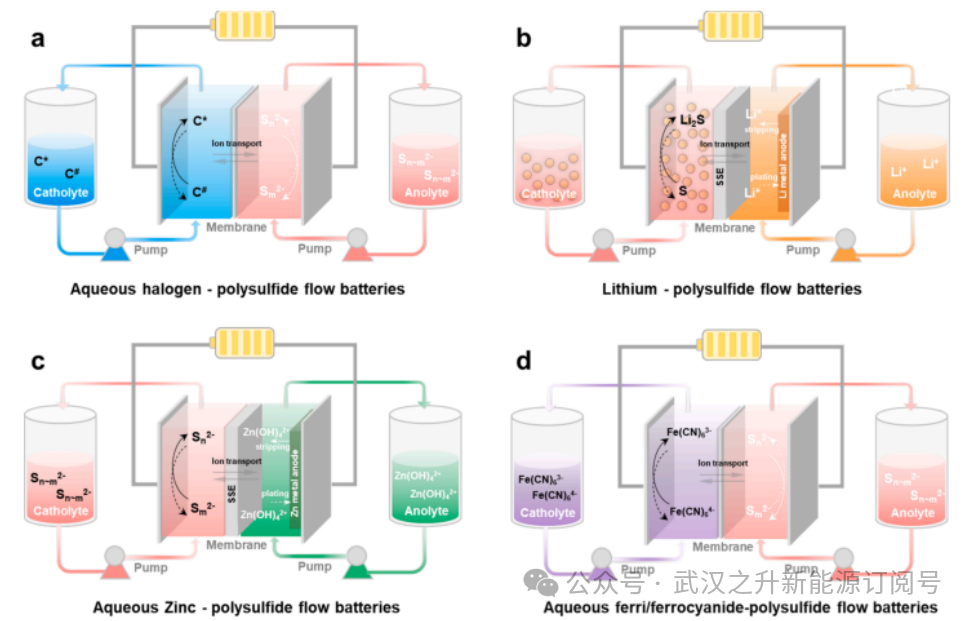
图1.不同类型的卤素-多硫化物水系氧化还原液流电池
硫具有理论能量密度高(1672mAh g⁻¹)、溶解度好、资源储量丰富、成本低廉且可靠性高等优势。如今,多硫化物已与多种阴极电解质材料结合使用,主要包括溴/溴离子、碘离子/碘酸根、锂/锂离子、锌/四氢氧化锌以及亚铁/亚铁氰化物。
1.1.水基卤素-多硫化物氧化还原液流电池
多硫化物基阳极电解质与溴阴极电解质配对使用时具有高开路电压。在阴极充电过程中,Br⁻被氧化为Br2并以Br₃⁻复合物形式存在;放电过程则相反。然而,溴的毒性会污染环境,存在重大安全隐患。与Br2/Br⁻体系相比,I⁻/I₃⁻体系具有更低的挥发性、更高的溶解度以及更快且高度可逆的动力学过程。然而,Br2/Br⁻或I2/I₃⁻在电解质中以复杂形式存在,对电极上的氧化还原反应动力学并无益处。此外,需要使用高碱性电解质来稳定多硫化物,与卤素氧化还原电对在中性或弱酸性介质中的偏好直接冲突,它们的电化学动力学性能更优。
1.2.金属-多硫化物氧化还原液流电池
锂-多硫化物液流电池或锌-多硫化物液流电池属于混合固液体系。以锂-多硫化物液流电池为例,通常使用纯锂金属电极作为固态电极。该体系存在枝晶形成问题,也是锂金属电池面临的主要挑战。此外,由于锂与水之间的相互作用,锂-多硫化物液流电池始终基于有机溶剂,尤其是对于锂金属负极而言。对于水性锂-多硫化物液流电池,锂金属负极与水性氧化还原活性物种正极之间由玻璃陶瓷膜分隔。然而,锂/水体系的一个主要缺点是需要无裂纹的玻璃陶瓷膜,该材料成本高昂(LICGC USD 90/cm²)且电阻率高(离子电导率低至1.0×10⁻⁴ S cm⁻¹)。对于水性锌-多硫化物液流电池,仍采用固态电解质作为阴极液与阳极液之间的隔膜。
1.3.水基铁氰化物/亚铁氰化物-多硫化物氧化还原液流电池
[Fe(CN)₆]³⁻和[Fe(CN)₆]⁴⁻具有明确的可逆氧化还原特性、高稳定性和低成本优势,是RFB极具潜力的正极材料候选物。Wang等人报道了一种铁氰化物/亚铁氰化物-多硫化物(Fe/S)氧化还原液流电池,正极采用高溶解度的碱金属铁氰化物/亚铁氰化物水溶液氧化还原材料,负极则使用碱金属多硫化物。Fe/S流系统采用相对环境友好的中性电解质,避免了使用强氧化剂、腐蚀性酸或溴等苛刻电池介质。然而,铁氰化物/亚铁氰化物阴极电解液的浓度通常较低(<0.8M),导致能量密度远低于已报道的RFB水平。但是,低溶解度极限的挑战可以通过阳离子调节策略来解决,并用作构建RFB的阴极电解液。
总之,在多硫化物基氧化还原液流电池中寻找最佳阴极电解质-阳极电解质组合仍是一项亟待解决的挑战。当前研究往往仅将特定材料(如膜、催化剂)随机应用于系统而缺乏系统性评估,为商业化应用提供了有限指导。
1.4.水基多硫化物氧化还原液流电池的局限性
基于硫的RFBs仍未实现广泛商业化应用,主要归因于多硫化物相关的三个持续存在的挑战:缓慢的氧化还原动力学、穿梭效应以及电解质不稳定性。首要挑战源于多硫化物电化学固有的缓慢氧化还原动力学,从根本上限制了基于多硫化物的RFBs的效率。在水溶液中,元素硫通过二硫键(−S–S−)断裂发生还原,依次形成短链多硫化物(Sn²⁻)和硫化物离子(S²⁻)。然而,水相硫氧化还原化学在Sn²⁻与S²⁻之间的转化过程缓慢,且受溶液中多硫化物复杂热力学效应的进一步影响。因此,电催化剂被广泛用于加速多硫化物氧化还原反应并提高电池效率。但电化学反应过程中水相环境中硫物种的具体形态仍未完全阐明。在传统的有机电解质基硫电池中,硫化物通常以稳定形式存在(如Li2S、Ma2S或K2S),且不会发生水解反应。而在水性电解质中,硫化物的形式(H2S(aq)、HS⁻和S²⁻)高度依赖于pH和电解质浓度等因素,显著影响硫化物反应的热力学和动力学。
第二个根本性限制源于现有膜对多硫化物渗透的选择性不足。多硫化物因其较小的离子尺寸和较低的电荷密度而易于在浓度梯度驱动下穿过膜。即使是阳离子交换膜(CEM)也常常无法阻止渗透。一旦迁移,多硫化物会与阴极电解液组分发生副反应,导致活性材料损失、容量逐渐衰减并最终引发系统失效。
第三个挑战涉及多硫化物电解质的内在稳定性。多硫化物物种间的转化反应会对氧化还原流电池的可逆性、实际容量及长期循环性能产生负面影响。
除了上述挑战外,在实际应用中还有一个关键但常被忽视的问题,容量衰减。缓解交叉反应导致的容量损失的一种成熟策略是采用对称化学体系,即两个半电池使用相同的母体活性物质。在此类系统中,交叉反应不会引发不可逆污染,且可通过定期电解液再平衡,即在各槽间转移并混合电解液以重新平衡活性物质浓度,来恢复电池容量。相比之下,基于多硫化物的电池通常采用不对称化学体系,每个半电池含有不同的活性物质;此时交叉反应会导致交叉污染及相应的容量损失,使得容量恢复变得极为困难。此类系统中电解液再平衡的技术可行性与经济影响仍知之甚少,相关研究也鲜有报道。因此,对于新兴的低成本液流电池要在大规模储能领域取得成功,必须超越材料改进层面,着力解决基础性的工程与运行挑战。
2.失败、缓解与恢复:构建可行的多硫化物基RFB的路径
2.1.通过电极与催化剂工程优化动力学性能
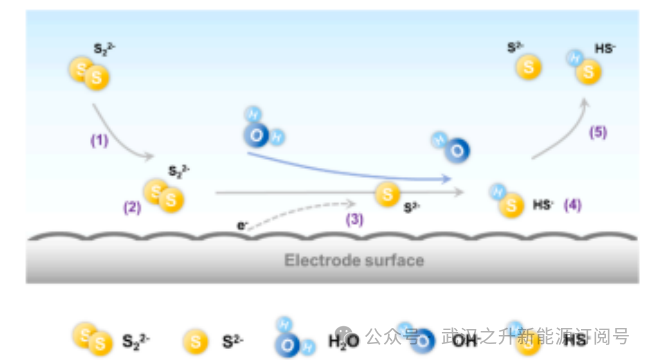
图2.电极表面多硫化物整个氧化还原过程的示意图
为克服多硫化物缓慢的氧化还原动力学所导致的显著极化效应及不良反应,必须优化具有高亲水性、电子导电性和催化活性的电极材料。在氧化还原液流电池中,电化学反应发生在电极/液态电解质界面。已知整个氧化还原过程包含多个基本反应步骤(图2):
(1)氧化还原物种从本体电解质向电极/液态电解质界面扩散;
(2)氧化还原物种在电极表面吸附;
(3)界面电荷转移;
(4)新形成的氧化还原物种从电极表面解吸;
(5) 新形成的氧化还原物种扩散回电解质。
2.1.1电极改性
适用于工业应用的电极改性技术主要分为两大类:热处理和酸处理。这些处理工艺的核心在于改变石墨毡的比表面积、表面活性位点、表面官能团及其结构。
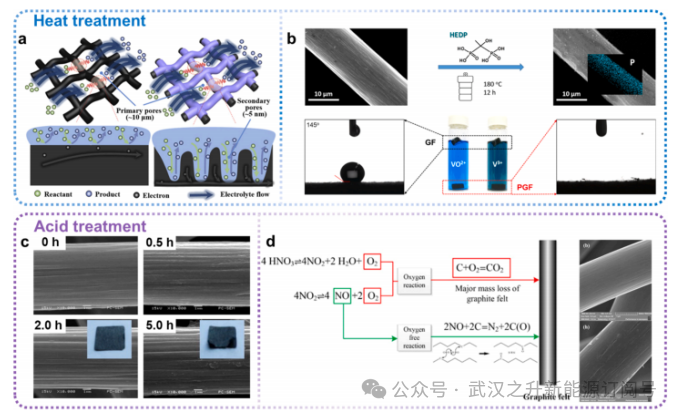
图3.电极改性策略
通过热处理可引入杂原子、含氧官能团并增加比表面积。为提高电解质的可及性,Zhou等人采用KOH作为蚀刻剂,在800°C下活化碳纤维,从而获得具有微孔和氧官能团的GF碳纤维(图3a)。热处理过程中碳纤维内部形成微孔,使其表面呈现粗糙多孔结构,从而提供更多活性位点。另一项研究聚焦于热氧化时长对电极结构与功能的影响:结果显示,随着氧化时间延长,原始碳毡的比表面积为0.187m²/g;经500°C加热6小时和9小时处理后的样品比表面积分别为4.353m²/g和5.746m²/g。Yu等人制备了磷掺杂石墨毡(PGF)作为全钒氧化还原液流电池电极(图3b)。该处理方法中,GF在空气中420°C加热10小时进行活化,随后浸入HEDP水溶液并在180°C下反应12小时。磷掺杂改善了GF的电解质润湿性,并在其表面诱导更多缺陷位点,显著提升了其对VO2+/VO2+及V²⁺/V₃⁺的活性与可逆性。
浓酸的液相氧化法被广泛应用于碳基材料的功能化处理,因其氧化程度可调、氧化效果优异且反应步骤简单。然而,较高的成本及废液无害化处理工艺复杂,导致工业应用效果差于热处理法。Yue等人开发了一种新处理方法:在聚四氟乙烯内衬的不锈钢高压釜中,在80°C条件下使用混合酸对碳纤维进行超声羟基化处理,实现了碳纤维的高效羟基化及处理后样品的高活性(图3c)。Wu等人提出采用硝酸蒸气在高温下处理石墨毡,经修饰的电极具有较大的比表面积和丰富的含氧官能团,可降低电极极化现象并提升电池整体性能(图3d)。总之,电极的热处理或酸处理是被广泛认可的改性策略,其中热处理更适用于大规模工业生产,且在电极的其他优化策略之前被广泛采用。
2.1.2电催化剂设计
为降低多硫化物氧化还原反应的能量势垒,电催化剂的设计需遵循以下关键要点:
(i)电催化剂需要具备高电导率和较大的表面积,以促进电子在电解质、电极和催化剂组成的三相界面之间快速转移;(ii) 电催化剂必须有效削弱硫-硫键,从而降低其断裂所需的能量势垒。为此,电催化剂应与硫化物物种发生相互作用。这种弱化作用可通过催化剂-硫键的形成实现,同时也有助于多硫化物在电极表面的吸附。在逆向反应过程中,催化剂-硫键会将不同的硫化物物种聚集在一起,从而促进硫-硫键的重构。
(ii)电催化剂需要在催化剂与硫的结合强度之间取得平衡。结合力过强会阻碍脱附过程并毒化活性位点,而结合力过弱则无法有效捕获电极表面的多硫化物。
电催化剂与多硫化物之间强烈的化学相互作用,对于PSRFBs中多硫化物的催化转化至关重要。过渡金属硫化物在电催化剂中尤为突出,因其具备促进体相或界面电子传输过程的理想特性:其带隙通常低于3eV,且表现出从半导体到金属级别的电导率。在PSRFBs中,常用的催化剂包括钴基、镍基和铜基硫化物。
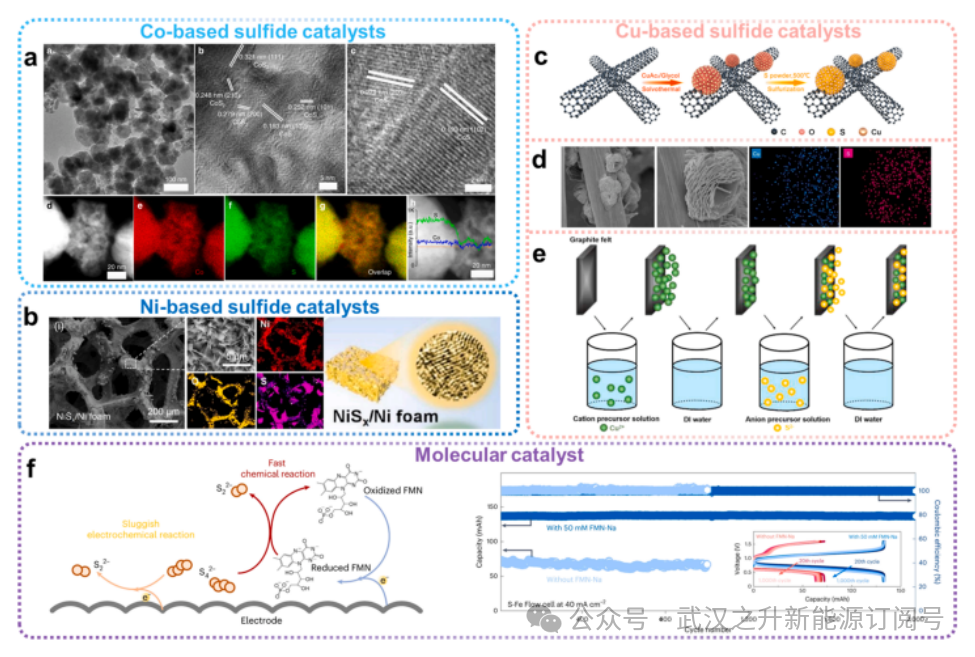
图4.催化剂改性电极策略
Liu等人设计了一种缺陷型二硫化钼纳米片负载钴单原子催化剂,该催化剂加速了S²–/Sₓ²–和I–/I3–的转化。原位实验与理论分析表明,钴单原子通过相变过程诱导二硫化钼中产生大量硫空位,增强了反应物及关键反应中间体的吸附能力并改善了电荷转移,从而提升了还原流电池性能。据报道,CoS₂/CoS异质结纳米颗粒(图4a)可在石墨毡上原位合成,通过提高带电离子的吸收率和促进电荷转移,增强S²–/Sₓ²–和I–/I3–氧的电催化活性。采用石墨毡负载CoS₂/CoS异质结的多硫化物/碘化物电池在10mA cm-2下可实现84.5%的高EE,连续运行约1000小时后EE保持稳定在96%。硫化镍催化剂也应用于多硫化物基液流电池:Jia等人开发了NiSₓ/Ni泡沫电极(图4b),对多硫化物离子具有电催化活性,并在使用S42-/S22-阳极液和MnO4⁻/MnO4²–阴极液的S/Mn还原液流电池中表现出优异性能。
相比之下,相对廉价的铜基催化剂无疑是更优选择。Qin等人提出Cu7S4/CNT复合材料可作为水性多硫化物-碘化物液流电池的双功能催化剂:Cu7S4表面的铜位点能有效稳定中间产物,从而促进电极反应;该复合材料(图4c)能高效吸附多硫化物和多碘化物并提升其电化学活性,最终实现水性多硫化物-碘化物液流电池高达84.6mW cm⁻²的功率密度。Mou等人开发了一种新型CuS纳米花修饰碳毡电极(CuS-CF)用于PFRFBs(图4d)。得益于卓越的电化学性能,CuS-CF复合电极在120mA cm⁻²下实现了68.9%的EE,显著优于原始CF电极。Wang等人报道了一种可行的连续离子层吸附与反应方法,可将硫化铜电催化剂接枝到石墨毡电极上,从而显著促进多硫化物的氧化还原反应(图4e)。采用该电催化剂的水性Fe/S液流电池在50mA cm⁻²下实现了77.7%的EE。除了金属硫化物电催化剂外,Lu等人(图4f)还开发出一种新型高效且耐用的分子催化剂。FMN–Na催化剂可将S-Fe RFB的过电位从>800 mV降低至30mA cm⁻²下的241mV。实验表明经催化处理的S-Fe液流电池在40mA cm⁻²下可稳定运行2000次循环,每循环衰减率仅为0.00004%。
尽管上述报道的催化活性在实验室规模下表现优异,但将其应用于工业PSRFB电堆面临三大实际障碍。首先,钴基催化剂的成本(约35–45美元/kg ¹)与PSRFBs的核心价值主张存在冲突;其次,催化剂负载方法必须保持碳毡电极的孔隙率和水力渗透性;在具有机械加工流道的实验室测试电池中,电解液被强制通过毡层,而在商业电堆中,电解液主要沿毡层流动,因此毡层的孔隙率对活性材料的接触至关重要;第三,在超过10000次循环的连续电解液流动条件下,催化剂的附着性能尚未得到验证,脱落的催化剂颗粒可能污染电解液并加速膜降解。
2.2.通过电解质工程实现稳定性提升
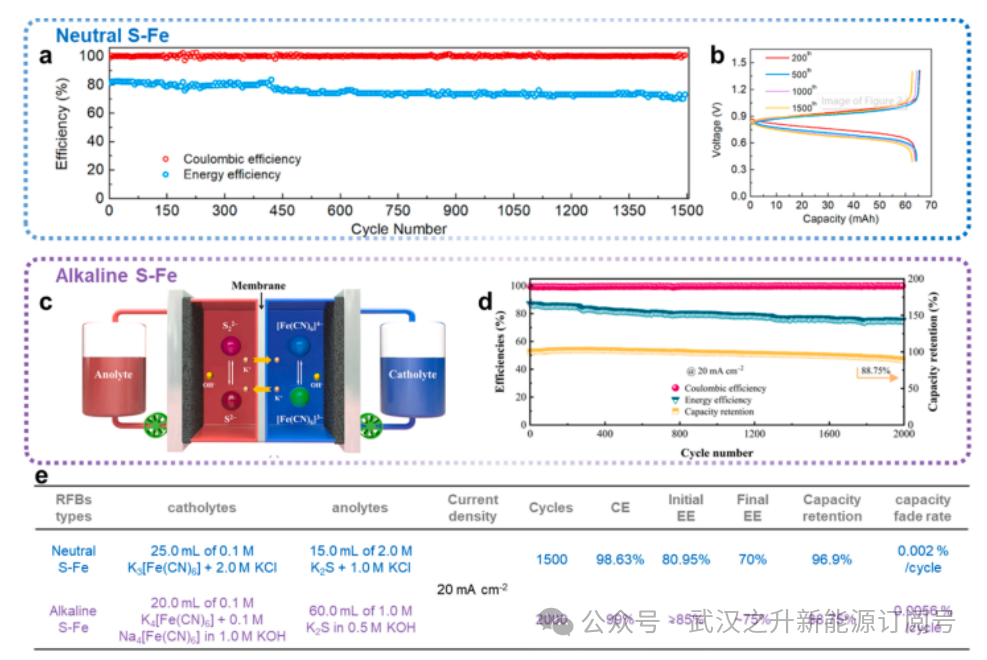
图5调节PH策略
从热力学角度看,多硫化物在水中倾向于发生水解,并产生大量HS⁻和OH⁻。由于水解作用,多硫化物溶液通常呈碱性。研究表明1M Na2S2和1M Na2S4溶液的pH值可达约13,水解平衡与pH密切相关:在酸性电解质(pH<7)中,平衡向右移动,HS⁻成为主要物种;更严重的是,低pH值甚至可能大幅增加有毒硫化氢的生成和释放风险,对PSRFBs构成灾难性影响,必须严格避免。而在碱性电解质(pH>7)中,随着pH升高,平衡向左移动,从而防止硫化氢析出及相关硫损失。因此,目前已开发出两种分别添加或不添加碱的PSRFBs。
Jia等人报道了一种中性S-Fe RFB(图5a、b)。这种中性S-Fe RFB表现出较高的容量保持率,在长时间循环测试后放电容量偏差极小。在低浓度下,1500次循环的容量保持率高达96.9%(相当于每次循环容量衰减率为0.002%)。在初始循环中,中性S-Fe氧化还原流电池的CE和EE分别为98.63%和80.95%。经过连续充放电运行后,EE在第1500次循环结束时缓慢降至70.00%。随后,研究人员还开发了一种碱性S-Fe RFB,电池采用KOH水溶液替代氯化钾水溶液作为支持电解质,如图5c、d所示。在相似浓度和电流密度条件下,碱性S-Fe RFB表现出较长的循环寿命,CE在2000次循环后仍保持>99.0%的高值。电池容量保持率为88.75%,相当于每循环衰减率仅为0.0056%。尽管EE(初始EE>85%)也有所下降,但在2000次循环后仍维持在约75%。尽管两项研究中的电解液浓度和使用方法并不一致(图5e),但仍可观察到:在碱性体系中,S-Fe RFB的EE得到了显著提升,表明副反应在一定程度上得到了抑制。然而,与碱性体系相关的其他若干问题也需谨慎考量,例如膜的稳定性、阴离子和阳离子的溶解度,以及HER。
2.3.通过添加剂和膜设计缓解穿梭效应
2.3.1添加剂
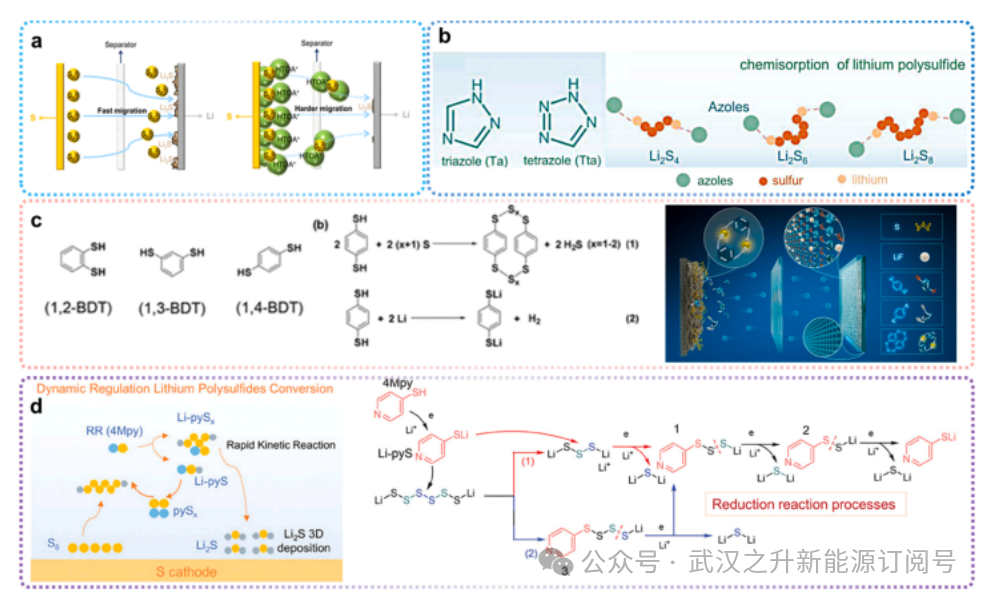
图6.添加剂策略
关于在多硫化物液流电池系统中使用添加剂来稳定电解液内多硫化物存在的研究报道较少,可以从其他基于水性多硫化物的电池或有机锂硫电池的相关研究中获得有益启示。Wang等人提出使用十六烷基三辛基碘化铵(HTOA-I)添加剂,在利用其较大尺寸的同时,保持多硫化物阴离子(Sn²⁻)的原始形态。大尺寸的HTOA ⁺与Sn²⁻的强结合抑制了多硫化物向锂金属负极的穿梭迁移,从而延缓了Sn²⁻的迁移(图6a)。Wang等人提出在锂硫电池中添加富含氮的唑类化合物作为电解质添加剂,例如三唑(Ta)和四唑(Tta)。三唑(Ta)和四唑(Tta)均含有多个氮原子,有利于LiPSs的化学吸附,从而有效缓解穿梭效应(图6b)。Lian等人提出了一类新型苯二硫醇(BDTs)电解质添加剂能与更多硫原子结合。如图6c所示,该化合物通过寡聚化与硫形成S-S键,从而改变硫的原始氧化还原路径。Kai等人提出在锂硫电池中使用有机硫化合物4Mpy作为电解质添加剂,该化合物可作为LiPSs的氧化还原调节剂,调控LiPSs的转化过程(图6d)。
上述非水系锂硫电池的实例具有参考价值,但由于存在三个根本性差异,将其直接应用于水系PSRFBs需要进行仔细重新评估。
(i)硫的形态:在有机电解质中,多硫化物以Li₂Sn形式存在且不发生水解。在水中,主要形态(HS⁻、S²⁻、Sn²⁻)具有pH依赖性,并可发生水解反应,会改变它们与添加剂的相互作用。
(ii)溶剂相互作用:高供体数溶剂能稳定锂硫电池中的自由基中间体。在水中,此类自由基寿命较短,因此添加剂设计应侧重于静电或路易斯酸碱相互作用,而非自由基稳定作用。
(iii)添加剂的化学稳定性:许多有机添加剂含有巯基或二硫键,这些基团在碱性水性电解质中可能被氧化或水解。未来研究应筛选水相兼容且碱性稳定的类似物,例如磺化或季铵化衍生物。
2.3.2膜改性
膜是RFB电堆的关键组件之一,其参数可进行定制以调控多硫化物泄漏。膜的厚度、当量重量及比溶胀系数始终会影响阴离子的渗透速率。除CEMs外,固体电解质也被报道可用于多硫化物基RFB。Martha M. Gross等人提出了一种“介导离子”策略:当前市售固体电解质中的Li⁺或Na⁺不直接参与阳极和阴极反应,而是作为被动的离子介体,促进电荷通过固体电解质传递。该介导离子型固体电解质已在多硫化物-多溴化物(PSB)和多硫化物-多碘化物(PSI)体系中进行验证,以消除氧化还原活性物质的交叉迁移现象。然而,陶瓷电解质在浓多溴化物和多碘阴极电解液中发生了腐蚀。此外,使用这类电解质会带来功率密度较低的代价。考虑到固体电解质成本高昂且集成工艺复杂,其在液流电池中的实际应用仍遥遥无期。对于工业应用而言,采用低成本方法对现有膜进行改性往往比开发全新的膜类型更具价值。
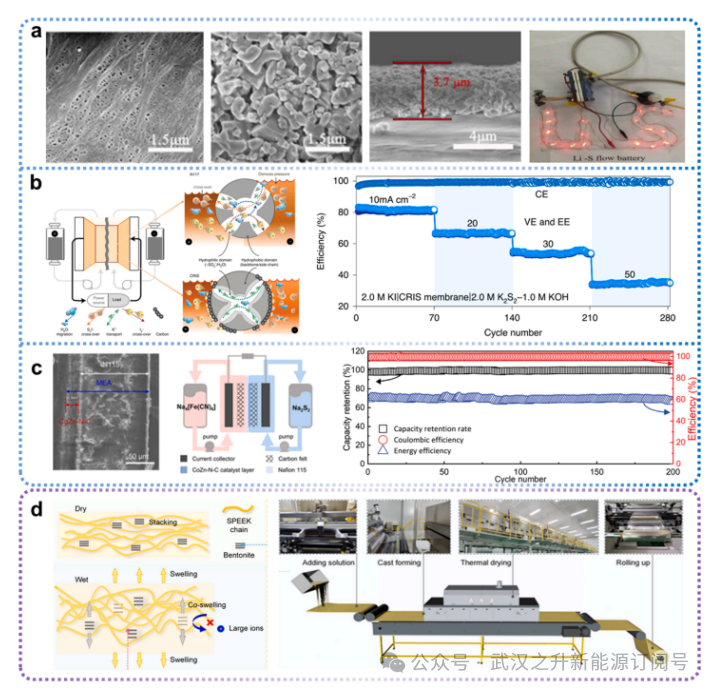
图7膜改性策略
在常用膜表面涂覆能够吸附多硫化物的材料,是抑制多硫化物穿梭核心结论应的有效方法。据报道,氧化铝颗粒被用于修饰聚丙烯隔膜(PP-Al₂O₃)(图7a)。与聚丙烯隔膜相比,PP-Al₂O₃隔膜表现出更好的热稳定性和电解质润湿性。密度泛函理论计算结果推断锂多硫化物与氧化铝之间可形成化学键。当PP-Al2O3隔膜用于Li-S RFB时,循环容量和倍率容量得到了提高,归因于极性Al2O3涂层不仅通过化学相互作用和物理屏障减轻了穿梭效应,而且通过良好的电解质润湿性促进了Li离子的迁移。Lu等人在Nafion膜上开发了一种由聚合物键合化学吸附碳构成的电荷增强层,用于吸附膜负侧的多硫化物阴离子和膜正侧的聚碘化物阴离子。本研究中,将PVDF结合的碳渗透到Nafion膜中。高疏水性的PVDF可减轻膜溶胀和吸水现象。高比表面积的Ketjen黑碳(KB)具有强大的多硫化物/聚碘化物吸附能力,能够捕获并积累带负电荷的阴离子,从而通过静电作用排斥电解液中的活性阴离子,降低离子交叉渗透。(图7b)然而,在相应的液流电池中,电荷增强型离子选择性(CRIS)膜表现出比N117更高的离子电阻和更低的VE,推测是由于碳层减缓了钾离子通过膜的传输。基于类似的涂层膜设计概念,Chen等人采用基于膜电极组件(MEA)的复合膜,为多硫化物还原流电池同时提供了催化效应和良好的渗透选择性。研究人员在N115膜上引入了基于MOFs的钴锌双掺杂N-C复合材料,以提升Na2S2的电化学吸附性能和氧化还原动力学(图7c)。该策略使得Fe-SRFB在60mA cm−2下,实现了超过99.7%的平均CE和200次循环后高达99.5%的容量保持率。
然而,涂层膜带来了对本已昂贵的Nafion™进行改性的额外成本,对整个行业而言可能难以接受。因此,未来在采用这些膜改性策略时,应优先评估成本更低的膜材料,例如非氟化烃基膜或国产全氟化膜。Chao等人通过在磺化聚醚醚酮(SPEEK)基质中引入膨润土作为共溶胀纳米海绵,开发出一种同步溶胀膜(CSM),从而构建出可动态调节离子筛分性能的纳米通道,有效解决了离子交换膜中众所周知的溶胀-选择性权衡问题。该CSM在碱性锌/铁电池中经过1300次循核心结论核心结论环测试,实现了95.88%的容量保留率。为应对大规模膜制造面临的挑战,研究团队开发了适用于CSM膜生产的中试规模合成及卷对卷制备工艺。该放大生产方法基本沿用了实验室制备流程,但针对工业应用进行了优化,实现了每平方米12.72美元的卷对卷生产成本(图7d)。
简而言之,膜改性工艺必须遵循确保可扩展性、成本效益及高通量生产的基本原则。此外,应更加关注实验室生产与工业生产之间的差异,因为膜的制备工艺会显著影响其厚度、等效重量和机械强度等关键性能。
核心结论
当基于多硫化物的RFB被大规模化并长期运行时,不可逆容量衰减仍是不可避免的挑战。因此,针对实用型PSRFBs的开发提出以下建议:
(1)契合市场需求并发挥独特优势:在当前发展阶段,多硫化物基RFB尚不具备与LIB在通用储能领域直接竞争的能力。因此,商业化策略应着重发挥其固有优势:能量与功率的独立可扩展性、较长的预期循环寿命以及低廉的原材料成,使其特别适用于LDES。未来的技术研发必须持续突出并优化这些核心价值主张。
(2)桥接实验室规模与工程系统在催化剂设计中的差距:催化剂的研发必须超越单纯的活性指标,解决实际工程中的限制因素:(i)催化剂必须具备高选择性以抑制副反应,特别是在碱性介质中的HER;(ii)必须严格评估催化剂负载量对碳毡电极孔隙率和水力渗透性的影响,尤其需考虑小型实验室反应池与大型商业化电堆之间流场动力学的差异;(iii)催化剂与碳基底之间需形成牢固且持久的附着,以防止长期运行中催化剂脱落和性能衰减;(iv)合成与负载工艺应优先考虑成本效益与操作简便性,以确保大规模生产的可扩展性。
(3)适用于规模化生产的膜改性设计:改性策略应与工业级卷对卷制造工艺兼容,以确保生产可扩展性、成本效益及高产量。此外,从实验室规模的膜制备向工业化生产过渡需谨慎考量,因为成型工艺(如浇铸、固化)的变更会显著影响膜的关键性能参数,例如厚度、等效重量及机械强度。
(4)优先开发多功能添加剂与电解质配方:引入功能性添加剂为电解质调控提供了便捷途径,通常能同时实现多项改进。添加剂可通过结合金属-多硫化物复合物、参与氧化还原反应或促进新反应路径的形成,从而有效抑制多硫化物穿梭现象。
(5)加速实用容量平衡技术的研发:缺乏可靠的容量再生方法,是阻碍PSRFBs实现商业化的关键障碍。为确保长期运行稳定性,并防止现场应用中因容量衰减导致系统故障,必须建立成本效益高且实用的容量平衡技术。
总之,尽管基于多硫化物的液流电池研究仍在持续进展,但该领域正处于关键转折点。传统方法虽获得了宝贵见解,但尚未开发出具有商业可行性的系统。本综述认为,未来的发展路径需要根本性的视角转变:从材料优化转向系统故障缓解,从实验室规模的性能指标转向工程规模的可制造性,从将容量衰减视为普遍现象转变为将容量恢复能力作为设计的核心要求。