
第一作者:强也
通讯单位:清华大学
通讯作者:赵雪冰
【成果简介】
以空气为氧化剂在低温下高效氧化5-羟甲基糠醛(HMF)制备2,5-呋喃二羧酸(FDCA)仍然具有挑战性。受电子载体介导的活细胞呼吸电子传递链(ETC)的启发,清华大学赵雪冰团队成功构建了人工ETC,并将液流燃料电池(LFFC)转化为柔性反应器,在温和条件下高效氧化HMF以产生FDCA。该LFFC反应器采用电沉积改性的泡沫镍作为阳极以促进HMF氧化,并采用(VO2)2SO4作为阴极电子载体以促进电子传递到空气中。通过选择阳极催化剂、调节外部负载和改变阴极电子载体或氧化剂,可以容易地控制反应速率。室温下的最大功率密度为44.9 mW cm–2,而对于FDCA的生产,短路条件是实现电子快速转移的首选条件。对于初始HMF为0.1M的单批操作,FDCA得率可达到97.1%。通过补料分批操作,FDCA浓度累积可达到144.5 g L–1,总得率为96%。Ni2+/Ni3+氧化还原对是介导电子转移的活性物种,实验和DFT计算结果同时表明HMFCA途径是优选的反应机制。相关成果以“Transforming liquid flow fuel cells to controllable reactors for highly-efficient oxidation of 5-hydroxymethylfurfural to 2, 5-furandicarboxylic acid at low temperature”为题发表在国际知名期刊Journal of Energy Chemistry上。
【研究背景】
2,5-呋喃二甲酸(FDCA)已被美国能源部列为最有前途的高附加值化学品之一,可作为生产生物基塑料聚2,5-呋喃二甲酸乙二醇酯(PEF)的单体替代石油基单体对苯二甲酸(PTA)。FDCA通常通过其前体5-羟甲基糠醛(HMF)的氧化产生。近年来,HMF氧化生产FDCA备受研究者关注,其中化学氧化似乎是最有潜力大规模应用的。然而,在化学氧化过程中,通常需要使用Pt、Au和Ru等贵金属催化剂及高温高压条件,而苛刻的条件通常会导致更高的成本和HMF的显著副产物形成。此外,常用的非贵金属主要包括Mn、Co、Ce和V,非贵金属催化剂虽然成本低廉、可用性高,但催化剂的制备通常复杂而耗时,并且HMF氧化的反应时间较长,生产效率较低。电氧化属于在外部电能输入下,在温和条件下实现HMF氧化的有效方法。该过程不仅可以避免苛刻的条件,而且通过调整氧化电位和电催化剂的选择,使氧化更容易控制。与大多数非均相化学氧化过程类似,电化学转化过程也涉及活性位点和电解质界面之间的表面反应,与化学氧化过程类似,尽管贵金属催化剂可以为HMF氧化提供更低的阳极电势,由于价格优势,开发非贵金属催化剂受到越来越多的关注,对非贵金属基催化剂,特别是钴和镍基催化剂对HMF氧化具有显著的电催化活性,受到了广泛深入的研究。然而,催化剂的制备过程较为复杂,如在严格的气氛下煅烧、在高温高压下的水热反应,甚至多重反应的偶联。因此,需开发一种简单有效的镍基催化剂制备方法。电氧化需要外部能量输入,此外,大多数电化学催化系统都在低HMF浓度下运行,因为较高的HMF浓度会增加腐殖质生产从而降低FDCA得率。因此,迫切需要开发更高效的生产方法,在温和的条件下,尤其是以空气为最终氧化剂,实现HMF的高效氧化获得高的FDCA得率和浓度。热力学上,由于氧具有1.229V的高标准电极电势,远高于羟基和醛基的电势,氧对HMF的氧化可以自发发生,但是氧化HMF的动力学速率低,导致在温和条件下转化效率较低。事实上,在自然界中生物体采用巧妙的设计,在自然条件下通过空气实现基质的氧化,如呼吸电子传递链(ETCs)作为一种线粒体途径,其中电子通过一系列含有辅酶或黄素蛋白作为电子介质的级联复合物,在1.1V的氧化还原跨度内从NAD+/NADH移动到O2/H2O。因此,受呼吸ETC工作原理的启发,本项工作旨在开发一种人工ETC,以促进电子从HMF转移到空气的动力学。特别地,本工作将液流燃料电池(LFFC)转化为HMF氧化反应器,其中HMF在阳极上转化为FDCA,而空气在阴极上得到还原。电沉积修饰的泡沫镍被应用为促进HMF氧化的阳极。以HNO3催化下的VO2+/VO2+为阴极电子载体,提高了空气(氧气)的还原效率。这种ETC能够在室温下,以O2为最终电子受体,以较高的FDCA选择性和得率快速氧化HMF。这项工作的目的是通过开发LFFC反应器,最大限度地提高FDCA的得率,并解释HMF在镍基阳极上氧化的机理。
【主要内容】
1. 用于生产FDCA和产电的LFFC体系的构建和原理
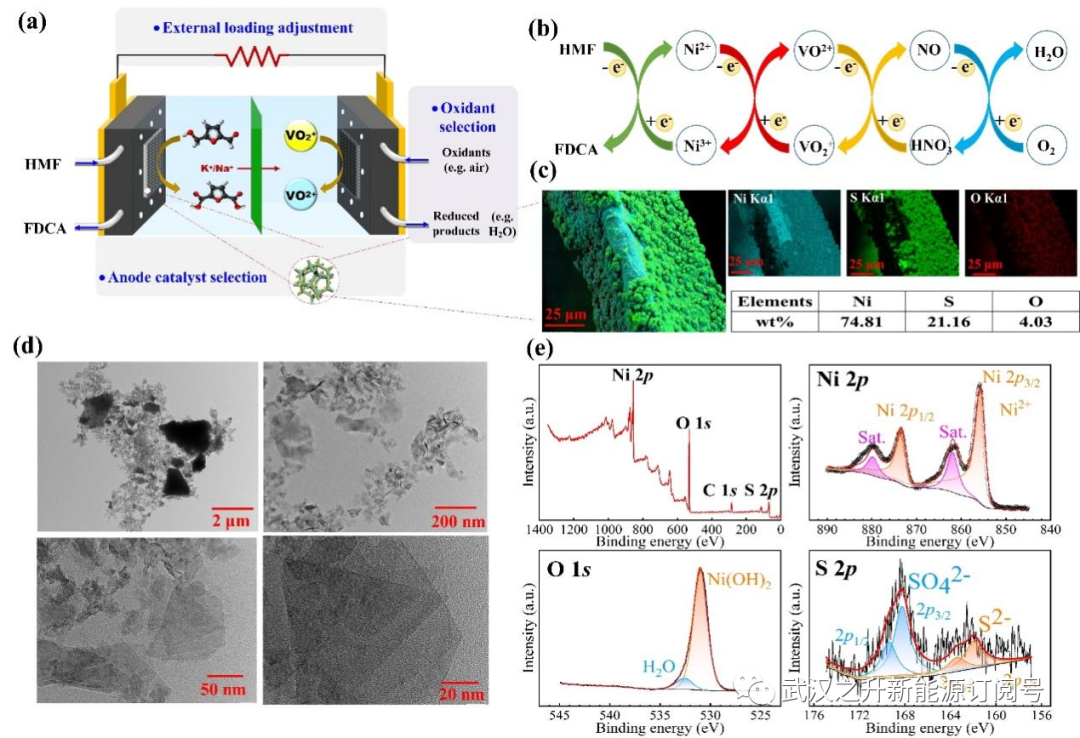
图1. 液流燃料电池(LFFC)反应器中HMF转化为FDCA的工作原理。(a):工作原理示意图;(b)以空气作为最终氧化剂构建的电子传递链;(c)Ni-S@NiF阳极的EDS图谱和元素分布;(d)Ni-S@NiF阳极的TEM图像;(e)Ni-S@NiF阳极的XPS全扫描光谱及Ni2p、O1s和S2p高分辨率光谱
本文所使用的液流反应器由武汉之升新能源有限公司提供


该体系的工作原理如图1所示。通过将LFFC转化为用于HMF氧化的反应器,可以通过热力学和动力学容易地控制该过程。HMF作为“燃料”被供给以释放电子,而空气(氧气)在电子载体的帮助下在阴极上被还原,从而在温和的条件下实现FDCA的生产(图1a)。Ni用作阳极催化剂,而硫酸氧钒((VO2)2SO4)用作阴极电子载体以接受从外电路转移的电子。当存在外部负载,同时实现电能输出。如图1b所示,通过ETC的构建促进了电子传递的动力学。在这项工作中,Ni-S复合物被电化学沉积在镍泡沫上作为阳极催化剂,以促进HMF的氧化,具体反应如下:
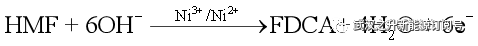
而在VO2+/VO2+和HNO3/NO氧化还原对的协助下,HMF释放的电子有效地传递给空气中的氧:
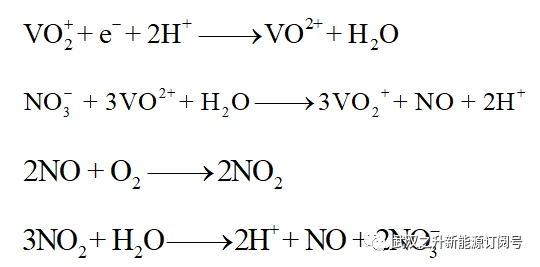
此外,在本项工作中,作者使用了碱性阳极电解液和酸性阴极电解液。这种不对称的酸碱设计可以很好地增加电子转移的驱动力,因此,整个反应本质是HMF被氧气氧化:
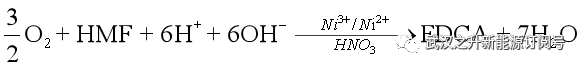
2. Ni-S@NiF阳极的制备

图S4. 通过CV60循环制备的Ni-S@NiF阳极TEM(a)和XRD(b)图像
通过循环伏安扫描电沉积60个循环制备得到具有最佳催化活性的阳极Ni-S@NF。从的SEM图像中观察到Ni-S@NiF阳极的Ni泡沫骨架结构没有改变,但基体表面被大量颗粒完全覆盖(图S4a),颗粒呈球形团簇。能谱(EDS)元素图谱显示,表面被74.8%的Ni、21.2%的S和4.03%的O覆盖。然而,在更大的放大倍数下,这些团簇似乎是由一些薄片而不是单个球体组成的。透射电子显微镜(TEM)图像(图1d)显示,球形颗粒实际上是由具有不同层的纳米片堆叠而成的,显著增加了表面积。Ni-S@NiF阳极的Ni2p、S2p和O1s高分辨率XPS光谱(图1e)与EDS结果一致。基于Ni2p高分辨率光谱,在855.67和873.40 eV处有两个主峰,分别对应于Ni2p3/2和Ni2p1/2,这归因于二价镍物种的存在,在861.90 eV和879.76 eV处的峰是相应的卫星峰。从O1s观察到的特征峰表明,镍在表面上的存在形式为Ni(OH)2。531.00和532.48eV处的峰分别对应于羟基氧和吸附的水。对于S2p XPS高分辨率光谱,161.93eV和163.41eV处的弱响应信号对应于S2p3/2和S2p1/2,而168.24和169.39eV的峰值意味着出现高氧化态S。然而,图中的X射线衍射(XRD)图S4b显示了对应于金属镍的(111)、(200)、(220)、(311)和(222)五个平面的衍射峰,这表明沉积在Ni泡沫上的Ni-S是无定形的。因此,通过CV电沉积的方法,在泡沫镍上沉积了大量的无定形的、硫掺杂的镍纳米片,形成了阳极催化剂(Ni-S@NiF)。
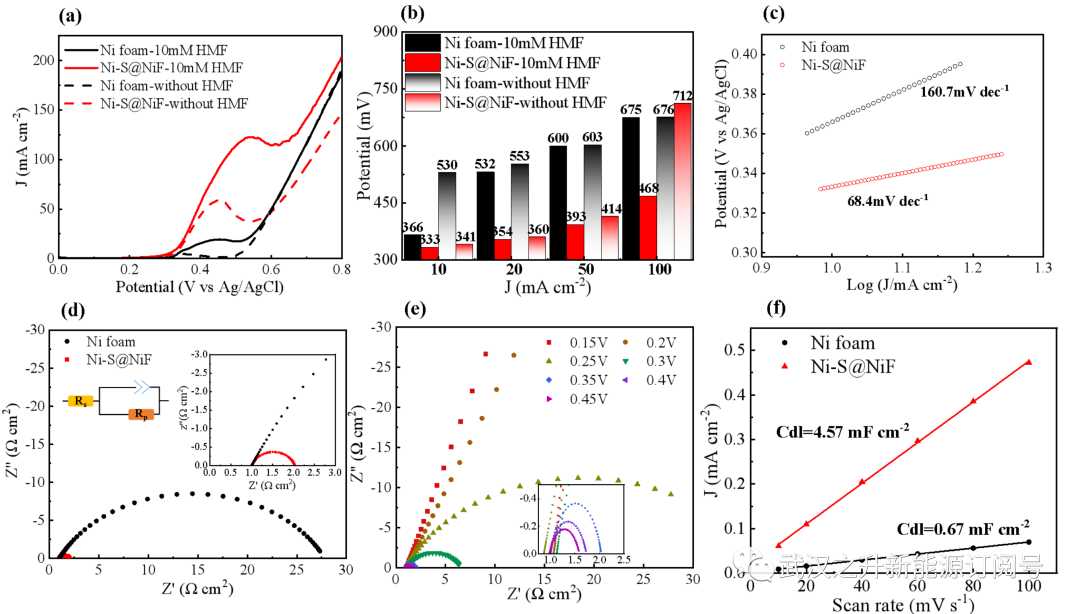
图S5. 制备的Ni-S@NiF阳极在1M KOH中电化学性能。(a)线性扫描伏安法(LSV)曲线;(b)使用10或100 mM HMF实现基准电流密度(10、20、50、100 mA cm−2)的过电势的比较;(c)Tafel曲线;(d)不同催化剂阻抗谱对比;(e)在100mM HMF的不同电势下的在阻抗谱对比;(f)双电层电容法测定电化学活性面积
为了进一步探索催化剂对HMF氧化(HOR)和析氧反应(OER)的催化活性,对Ni Foam进行电沉积改性前后的电化学性能 (图S5)。在10mM HMF的存在下,Ni-S@NiF阳极的HOR贡献的电流密度显著增加(图S5a)。使用Ni-S@NiF达到一定电流密度(例如10、20、50和100 mA cm-2)所需的电势明显低于未改性的泡沫镍(图S5b),这表明Ni-S@NiF电极具有优异的催化性能,并能有效降低HOR的过电位。Ni-S@NiF阳极的Tafel斜率(68.4 mV dec-1)小于Ni泡沫阳极的一半(160.7 mV dec-1),这意味着Ni-S@NiF阳极具有更高的HMF氧化反应速率(图S5c)。通过电化学阻抗谱(EIS)评估了电极界面的电子转移过程。显然, Ni-S@NiF在低频区域的半圆半径小得多(图S5d)。并且由于通过搅拌和添加100mM HMF消除了底物扩散限制,在高频区域中没有观察到线性区域。通过等效电路拟合,得到了了溶液电阻(Rs)和极化电阻(Rp)。尽管Rs几乎相同,但Rp从26.83Ω显著降低到1.03Ω,表明HMF在Ni-S@NiF电极表面上的氧化电子转移速率更快。此外,不同电势下的阻抗谱(图S5e)显示,在低电位下没有观察到半圆,半圆半径随着施加电势的增加而变小。当电位增加到0.25V时,Rp突然降低到36.02Ω,这意味着在低于0.25V的电位下几乎不可能发生HMF氧化反应。电化学活性面积(ECSA)通过双层电容(Cdl)的方式进一步确定,结果表明电沉积显著提高了ECSA,有利于提高HMF氧化的催化效率。因此,Ni-S@NiF阳极对HMF氧化显示出更多的电化学活性位点和更快的电子转移速率,显示出优异的HMF催化活性。
3. 在LFFC中通过HMF的阳极氧化生产FDCA
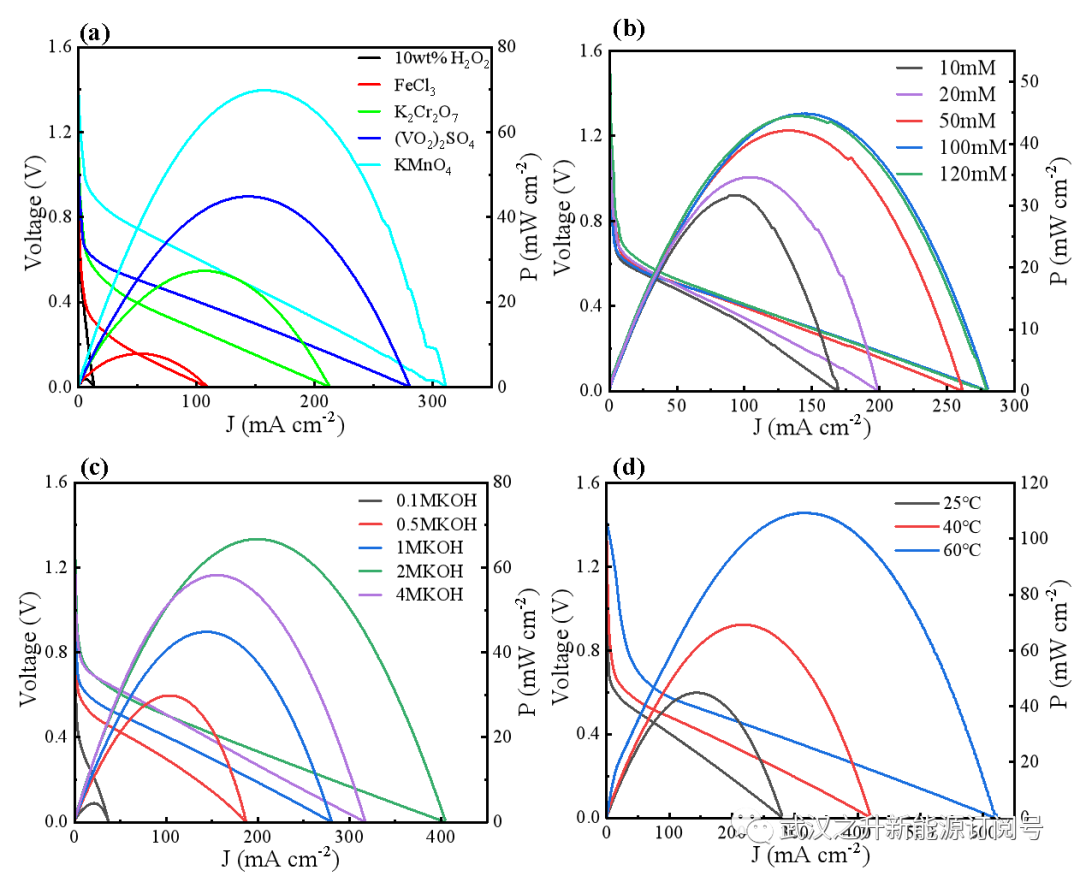
图S6. 不同工作条件下LFFC功率密度的比较。(a)阴极电子载体; (b) HMF浓度; (c) KOH浓度; (d)放电温度
LFFC反应器最重要的优点之一是氧化和还原反应分别发生在阳极和阴极上,避免了氧化剂与产物FDCA之间的直接接触,有利于后续的回收和纯化过程。由于LFFC将化学能转化为电能,因此放电性能也能很好地反映电子转移速率。为了更高效的生产FDCA,进一步研究和比较了不同操作参数(阴极电子、底物浓度、碱浓度、温度,外接负载)对电池性能和FDCA的合成的影响(图S6–S7)。通常,为了减少腐殖质的形成并增加FDCA得率,优选短路放电以实现高的电子转移速率,通过改变反应温度、阴极电解液中氧化剂,阳极液底物浓度、碱浓度,可以调节电子转移的动力学,调控反应过程。结果显示通过改变放电条件确实可以很容易地控制HMF的转化率和FDCA得率,最终优化得到了最佳的反应条件为,在室温下短路放电,以硫酸氧钒为阴极电子载体,使用1M KOH碱液并添加100mM HMF时为优选条件。
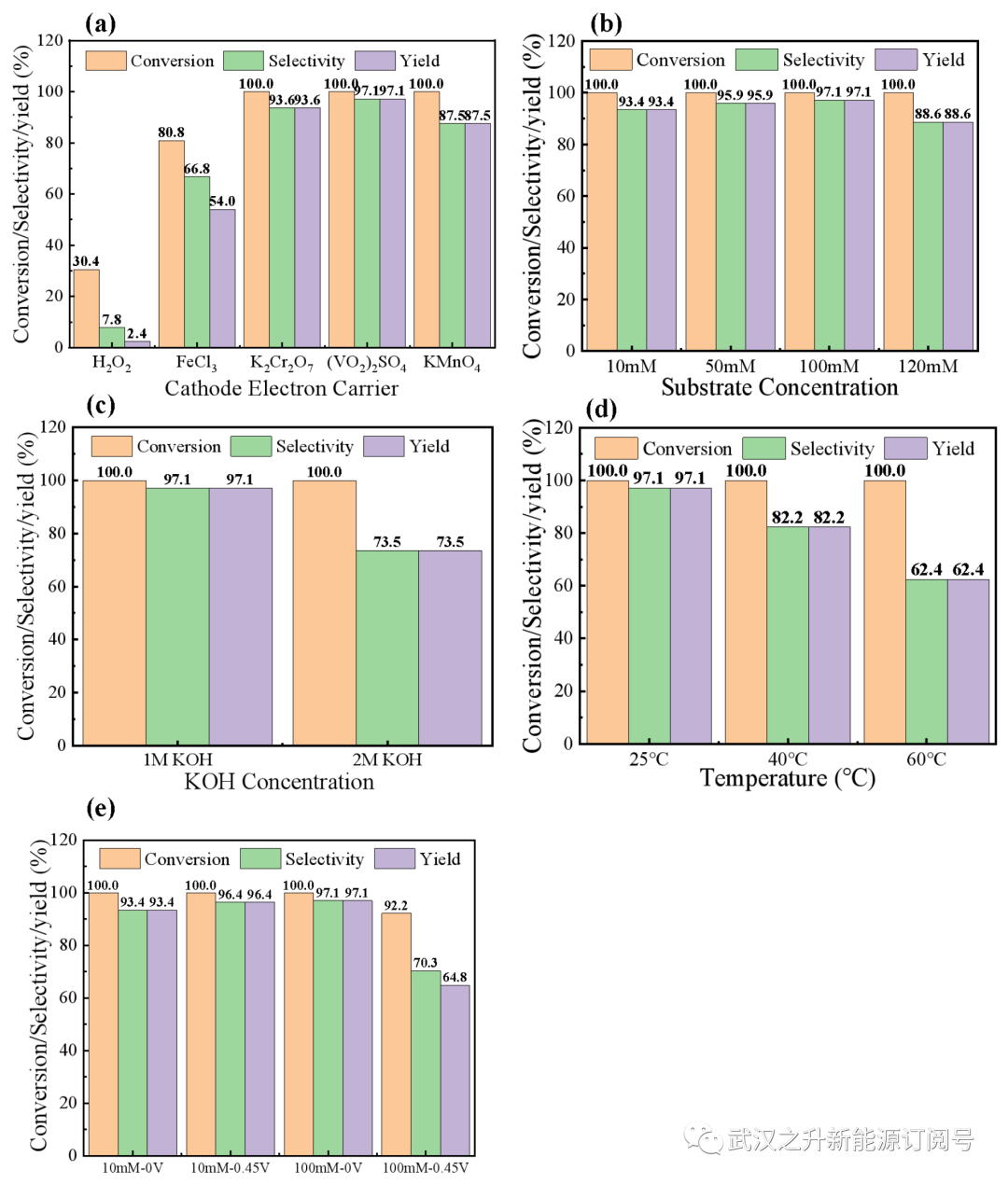
图S7. 不同操作条件下HMF转化率、FDCA选择性及得率。(a) 阴极电子载体; (b) HMF浓度; (c) KOH浓度; (d)温度; (e)输出电压
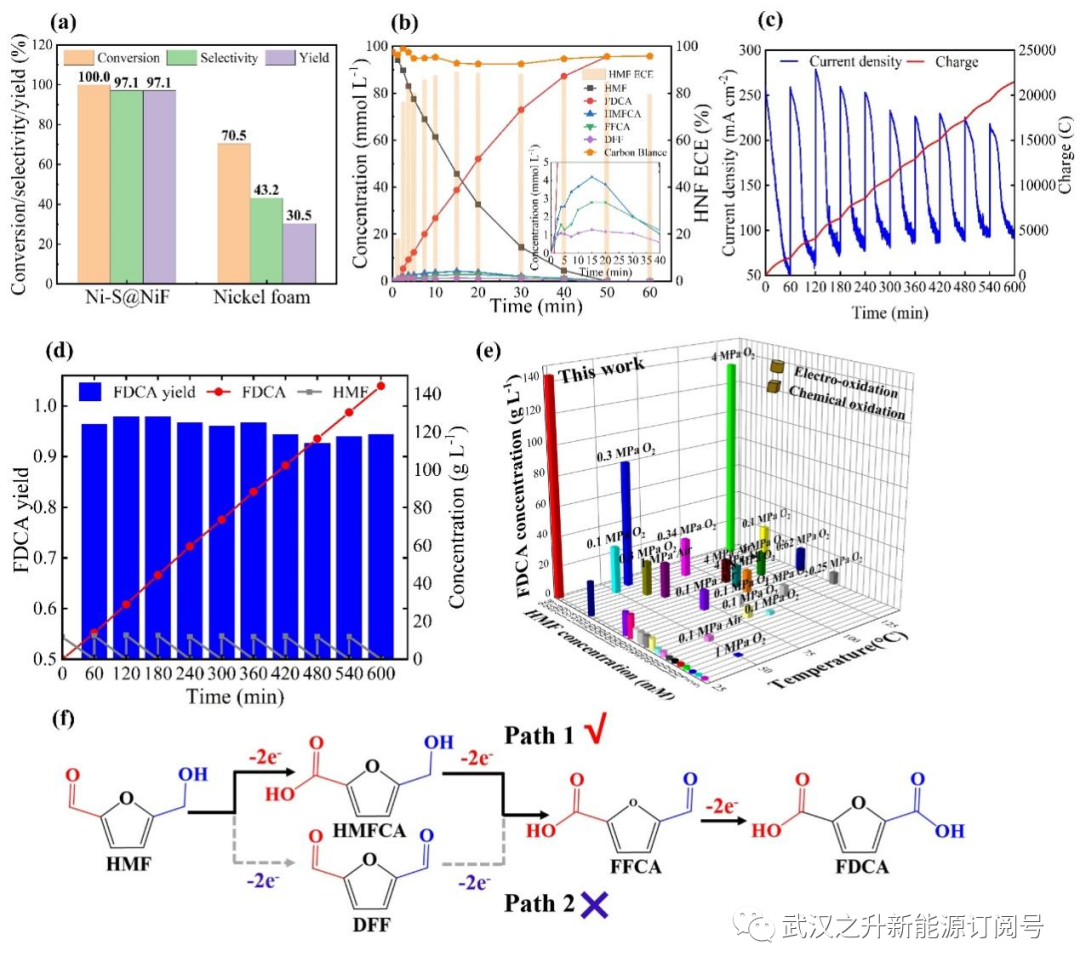
图2. 开发的LFFC反应器用于生产FDCA。(a)Ni-S@NiF和泡沫镍阳极的比较;(b)放电期间的产品分布;(c)分批补料操作期间的电流密度及FDCA浓度的变化;(d)分批补料操作期间过程中的FDCA得率和浓度;(e)本工作获得的FDCA浓度与已有报道的比较;(f)在LFFC反应器中HMF氧化形成FDCA的两种可能途径
与原泡沫镍相比,以Ni-S@NiF作阳极可以获得明显更高地HMF转化率和FDCA得率(图2a),表明Ni-S的电沉积修饰可以很好地改性和活化泡沫镍,提高HM的氧化速率。产品分布如图2b所示,在放电过程中,HMFCA和FFCA主要积累。通常HMF氧化为FDCA有两种途径,如图2f所示,上述结果表明,在LFFC反应器中,HMF的氧化主要通过HMFCA途径进行(途径I)。众所周知,醛(-CHO)在碱性介质中表现出比羟甲基(-CH2OH)更高的反应性,HMFCA途径是本工作中LFFC反应器阳极上HMF氧化的首选机制。通过长时间的分批补料放电,以研究阳极的稳定性并提高阳极FDCA浓度。如图2c所示,电流密度随着放电时间增加而降低,但当加入HMF时,电流密度突然增加,同时转移的电荷量不断累积。HMF在每次补料中都能快速氧化,伴随随着FDCA浓度的增加。10批次补料过程中,FDCA每次得率均保持在93-98%,FDCA累积浓度可达到144.5 g L–1(0.93 mol L–1),总得率为96%(图2d),有利于后续的分离纯化。与一些在不同HMF浓度和反应温度下,HMF的化学和电化学氧化以产生FDCA的结果相比,本工作产物浓度处于最高水平(图2e)。通过补料分批操作,可以获得更高的FDCA浓度,并且平均得率高达96%。此外,本项工作中的LFFC反应器可以同时发电,而不是消耗外部电能。通过酸化、沉淀、洗涤和再结晶,可以容易地回收和纯化所获得的FDCA。干燥后可获得纯度为99.9%的白色粉末。因此,开发的以为Ni-S@NiF阳极的LFFC反应器,在氧化HMF生产FDCA方面表现出优异的性能,具有潜在的环环保和经济前景。
4.LFFC反应器中阳极氧化HMF电子转移的机理探讨
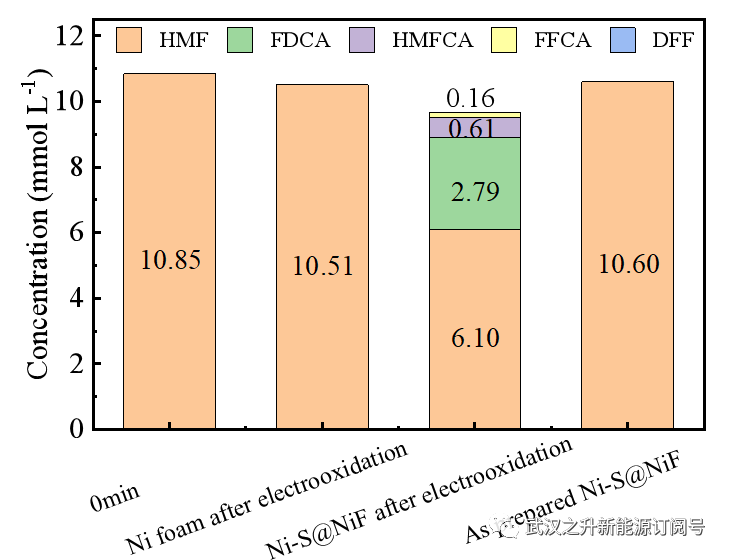
图S14. 开路(不放电)并在碱性条件下,不同阳极处理HMF所得产物分布
由于阳极上只电沉积了Ni,因此金属镍一定是HMF氧化的活性中心。先前的研究表明,在高电位的驱动下,Ni3+有利于以NiOOH的形式存在于碱性溶液中,这是实现HMF化学氧化的最重要的活性位点。为了验证这一推断Ni-S@NiF在1M KOH中在0.4V的恒定电势(没有OER)下被完全氧化。随后,将电氧化后的阳极直接浸入含有10mM HMF的1M KOH中,10分钟后通过HPLC监测产物的分布(图S14)。不出所料,只有电氧化的Ni-S@NiF阳极导致FDCA和一些中间产物的形成。然而,电氧化镍泡沫和新制备的Ni-S@NiF由于缺乏Ni3+物种,阳极不能实现HMF的氧化。
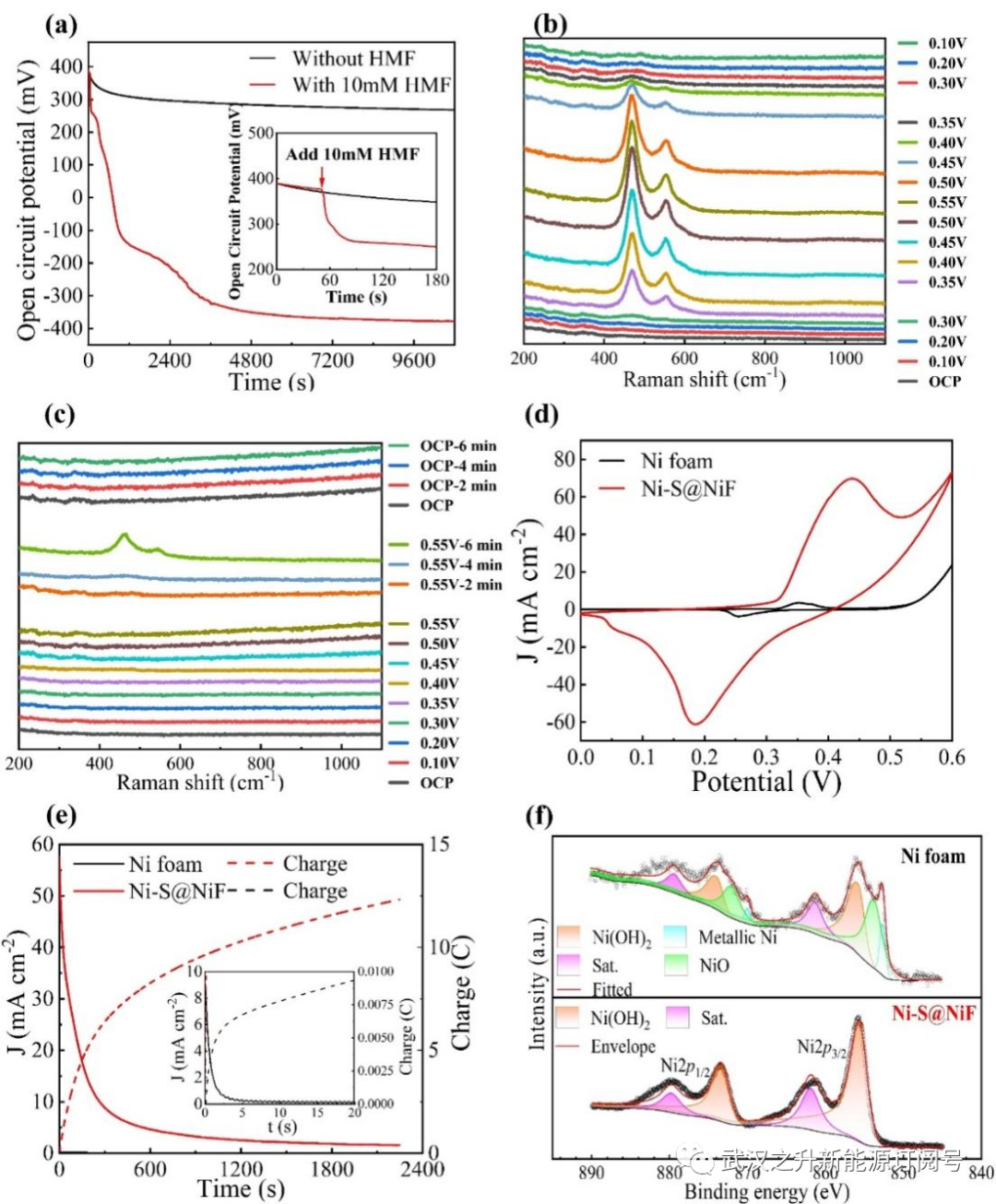
图3. Ni-S@NiF用于HMF氧化的阳极性能改善机制研究。(a)开路电位与时间的关系曲线;(b)1M KOH中的原位拉曼光谱;(c)在含有10mM HMF的1M KOH中的原位拉曼光谱;(d)在不含HMF的1M KOH中的循环伏安曲线;(e)在0.4V下对阳极进行恒电位电解,电流密度和转移电荷随时间的变化;(f) Ni 2p高分辨率XPS光谱的比较;
如图3a所示,研究了阳极在1M KOH中的开路电位(OCP)随时间的变化,以证实Ni2+/Ni3+对的化合价变化,由于从Ni3+到Ni2+的自发转变(NiOOH+H2O+e–→Ni(OH)2+OH–),在没有HMF的情况下反应进行缓慢。当向溶液中加入10mM HMF后,电位急剧下降,表明立即发生了NiOOH介导的HMF氧化(NiOOH+HMF→Ni(OH)2+氧化产物),并且最终稳定在更负的电位,这归因于HMF氧化对Ni3+的快速还原。进一步通过原位拉曼光谱测试,以证实Ni3+/Ni2+在Ni-S@NiF阳极表面的可逆转变(图3b–c)。对于OER,当电位从0.10 V增加到0.55 V时,约470和550 cm–1处的信号变得更清晰,这归因于Ni–O振动,表明NiOOH的原位形成。当施加的电势降低时,峰值逐渐减弱并最终消失表明Ni3+减少。相反,在存在10mM HMF的情况下,即使施加电势增加,也没有观察到特征峰,这表明HMF和NiOOH之间的化学反应很快NiOOH立即被原位还原。然后,探索在0.55V下连续电解6分钟,由于相对长时间的反应消耗了HMF,检测到了NiOOH对应的信号峰。然而,一旦移除电压,NiOOH的信号再次消失,这充分证明了NiOOH在阳极上自发氧化HMF(NiOOH+HMF→Ni(OH)2+氧化产物)。为了探索Ni2+/Ni3+电对的氧化还原特性,记录了循环伏安曲线(图3d)。在0和0.6 V之间存在一对氧化还原峰,分别对应于Ni2+和Ni3+的阳极氧化和阴极还原,对于Ni-S@NiF阳极,峰值强度明显大于泡沫镍的峰值强度,表明存在更多的活性的Ni2+。如图3(e)所示,在1M KOH中,0.4 V(不发生OER)的恒电位电解过程中,Ni-S@NiF阳极观察到更大的电流密度,表明在表面上存在更活性的Ni2+物种。此外,Ni 2p的高分辨率XPS光谱(图3f)显示,对于Ni-S@NiF阳极,表面存在大量的Ni(OH)2而不是单质镍和氧化镍。另一方面,Ni2+的结合能从855.84 eV负移到855.67 eV,表明局部电子密度增加,导致结合能降低,这更有利于Ni2+失去电子并转化为Ni3+。因此,通过对泡沫镍进行电沉积修饰,对于Ni-S@NiF阳极,实现了活性Ni2+物种数量的显著增加,电子结构的有益变化有利于NiOOH的产生,使泡沫镍得到显著的活化。
基于已有研究,本工作提出了一种可能的反应机理,如图4a所示,在强碱溶液中,反应由氢氧根离子对醛基羰基碳的亲核攻击引发,形成水合物阴离子中间体(二醇阴离子,GDA)。在OH−的帮助下,GDA中间体释放氢负离子,即脱氢,导致HMFCA的形成。释放的氢负离子自发转移到Ni-S@NiF阳极表面的NiOOH上,导致Ni(OH)2的形成。在阴极氧化剂(硫酸氧钒)的驱动下,电子释放到电极上,Ni(OH)2被再氧化形成NiOOH,可以进一步氧化HMF并转移电子。HMF分子的醇基通过α-C-H和羟基依次脱氢,共转移两个氢原子,进一步氧化为醛基形成FFCA。之后,FFCA经历类似的水合和脱氢步骤以形成最终产物FDCA。因此,整个过程包括通过NiOOH氧化醛和羟基,以及通过用阴极氧化剂氧化Ni(OH)2,再生NiOOH。值得注意的是,如原位拉曼光谱所揭示的,速率决定步骤是NiOOH的再生,这表明促进Ni2+向Ni3+的转化将是开发新催化剂或工艺以提高工艺效率的重要策略。
为了研究反应机理,通过构建NiOOH(001)模型进一步进行了密度泛函理论(DFT)计算。包括相应的中间体稳定吸附构型的自由能图如图4b所示,HMF到 HMFCA为自由能下降过程,而DFF合成需要大的能垒(1.25eV),这表明本工作中用于FDCA合成的DFF途径在热力学上不是优选的,而HMFCA途径更可行。这一结论与实验结果一致,即HMFCA浓度远高于DFF浓度。就HMFCA途径而言,由于最大能垒(0.99 eV),O–H键的断裂被确定为热力学速率决定步骤。因此,未来工作中的催化剂和工艺设计应侧重于促进HMFCA向FFCA的转化。
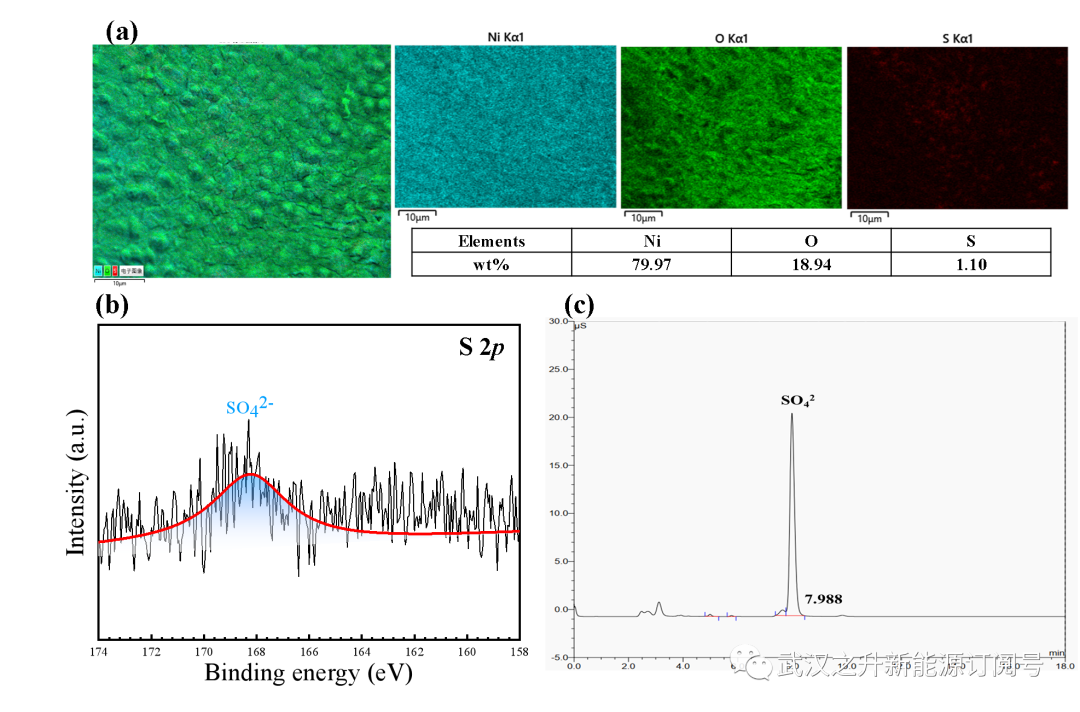
图S17. Ni-S@NiF阳极放电后的EDS (a)和XPS (b)图像; (c)电解液在1M KOH中放电后的阴离子色谱分析
为了研究电沉积过程中引入的硫元素的走向,观察了EDS和XPS图像,如图所示。S17(a–b)。可以清楚地观察到,硫元素在放电后从阳极表面消失。放电后对电解质的阴离子色谱分析表明,电解液中存在SO42-(图S17c),表明硫元素从阳极表面浸出并进一步氧化为SO42-,因为阴极氧化剂具有足够高的氧化电位氧化硫元素。因此,泡沫镍在电沉积修饰和放电过程中活化示意图如图4c所示。电沉积在泡沫镍表面引入了Ni和S,在1M KOH中阳极重构为Ni(OH)2,随后在放电过程中在阴极氧化剂的氧化下进一步转化为NiOOH。因此,由于离子半径的大小不同,硫的引入以及随后的浸出和氧化有助于形成更粗糙的几何结构,并且有利的电子结构提高了导电性,从而提高了催化性能。在HMF存在的情况下,NiOOH迅速被还原并伴随着FDCA的产生。在阴极侧,VO2+通过外部电路接收电子,最终在NO3–的催化下由O2再生。因此,本工作中开发的LFFC反应器中构建的电子传递链(图4d),可以在室温下用廉价的氧化剂(空气)连续氧化HMF生产FDCA。
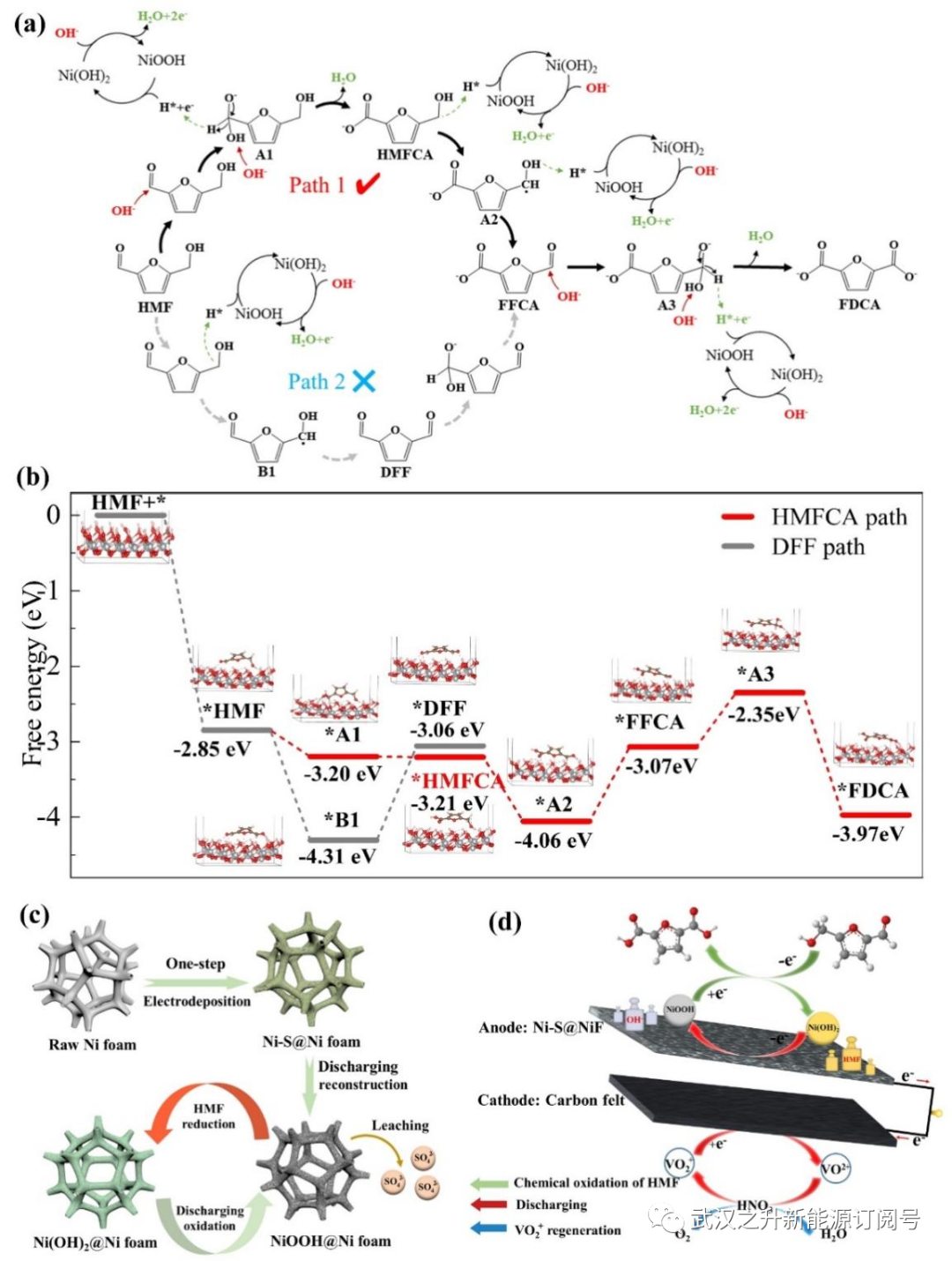
图4. LFFC反应器中Ni-S@NiF阳极将HMF氧化为FDCA的机制(a)HMF氧化的反应途径;(b)在NiOOH上HMF氧化中间体的自由能台阶图;(c)电沉积修饰和放电活化泡沫镍的示意图;(d)用于在LFFC反应器中,在温和条件下连续生产FDCA的电子转移链
【主要结论】
通过将LFFC转化为反应器,在低温下以空气作为最终氧化剂,实现了HMF的有效氧化以生产FDCA。通过引入Ni和S元素对泡沫镍进行电沉积修饰,制备的Ni-S@NiF阳极对HMF氧化表现出优异的催化活性。以(VO2)2SO4为阴极电子载体,在HNO3的辅助下实现电子向氧的转移。LFFC还使用了酸碱电解质的不对称设计来提高电子转移的驱动力。室温下的Pmax为44.9 mW cm−2,对于FDCA的生产,优选在短路条件下进行反应以实现电子的快速转移。通过补料分批操作,FDCA浓度可达到144.5 g L−1,得率约为96%。机理分析表明,Ni和S的电沉积对泡沫镍表面有很大的改性作用,HMF氧化的活性物质实际上是NiOOH,S在放电过程中被浸出和氧化,进一步活化了泡沫镍。Ni2+/Ni3+氧化还原对是介导电子转移的活性物种,实验和DFT计算结果同时表明,该LFFC反应器中的HMF氧化主要通过HMFCA途径进行。因此,将液流燃料电池转化为实现HMF氧化的反应器,可以通过选择阳极催化剂、调节外电阻和选择阴极氧化剂来容易地控制反应过程。
【文章引用】
Ye Qiang, Xi Liu, Denghao Ouyang, Zhao Jiang, Xuebing Zhao. Transforming liquid flow fuel cells to controllable reactors for highly-efficient oxidation of 5-hydroxymethylfurfural to 2, 5-furandicarboxylic acid at low temperature ,2023, Journal of Energy Chemistry
https://doi.org/10.1016/j.jechem.2023.11.023