
第一作者:Zhongxin Chen
通讯单位:新加坡国立大学和天津大学
通讯作者:Kian Ping Loh
【成果简介】
缺点:在多相催化中,长期以来的挑战是在液相有机转化中实现与均相或酶促过程相当的极高活性和化学选择性。开发具有原子精确配位的单原子催化剂(SAC)的目的是模拟均相途径,但由于金属浸出或聚集而面临稳定性问题。此外,由于缺乏标准的制备方案和使用实验室分批反应器的低转化率,SAC在化学生产中的实际应用受到阻碍。
本文重点介绍了作者最近在催化剂设计和反应器级工程方面通过SAC流动合成精细化学品的研究。在催化剂和反应水平上,作者将讨论SAC催化过程中控制反应性和化学选择性的内在机制,并重点介绍SAC优于其他催化方法的例子。具体而言,作者报道了SAC介导的药物的制备和后期功能化,包括依序或多组分方式通过化学选择性转化制备药物,包括山莨菪碱、达菲、卡沃松斯塔特、吲哚美辛和许多其他药物。强调了超高负载SAC为涉及两种或多种反应物的有机转化提供多位点途径的能力,并与传统SAC中的单位点途径进行了对比。使用操作X射线吸收光谱法获得的对动态催化循环的分子水平理解为设计更有效和耐浸出的SAC提供了指导。这也要求通过配方将实验室粉末基催化剂转化为工业上可行的整体催化剂,以进一步提高耐浸出性。在反应器层面,作者将强调连续流技术在最大限度地提高生产力和简化从实验室到商业生产的过程转移方面的重要性。特别讨论了使用燃料电池型流动堆定量生产精细化学品,包括使用Pt-SAC模块(3.2 mg Pt)以5.8 g h−1的生产率合成多功能苯胺。最后,作者还确定了SAC研究中需要克服的技术障碍,并就大规模生产的标准化和可扩展协议、典型SAC的生产成本分析、高通量筛选平台以及涉及光化学和电化学反应的新型流动反应器设计提供了作者的观点,以扩大SAC的化学生产规模。
【研究背景】
以节能和可持续的方式生产未来的化学品一直是催化研究的中心。具有原子分散活性中心的多相单原子催化剂(SAC)是非常理想的,因为它们具有最佳的原子利用效率和与均相催化中配体配位活性中心的结构相似性。这引起了人们对SAC介导的用于合成精细化学品的液相有机转化的兴趣。研究表明,由于多功能分子与特定官能团的优先相互作用,许多氧化和加氢反应对多功能分子具有显著的化学选择性。SAC在重要的交叉偶联反应中的应用,包括Suzuki、Ullmann、Sonogashira、Heck和Glaser偶联,以构建碳–碳键,以及最近的C−O和C−N交叉偶联反应,都有报道。这些反应通常涉及化合价的动态变化,氧化加成–还原消除途径中催化中心的状态; 因此,在催化剂设计中实现结构灵活性和耐浸出性之间的微妙平衡从根本上来说是具有挑战性的。SAC也已证明在复杂反应中是有用的,尽管这些反应迄今为止缺乏通用性。其中包括炔烃和烯烃的二穿孔和硼氢化、加氢甲酰化、氧化环化和烯烃异构化–氢化硅烷化,以生产各种有价值的构建块(例如硼酸衍生物)。SAC在促进这种复杂反应中的独特特性为药物的合成和后期功能化铺平了道路,包括洛林达明、达菲、卡沃松斯塔特、吲哚美辛、苯丙嗪、布洛芬、芬地林、西那卡司盐酸盐、和许多其他传统上通过均相工艺生产的药物。(图1A)。
然而,缩小到单原子水平给化学生产带来了一系列独特的挑战(图1B)。例如,尽管最近证明了一些特殊反应,但已知SAC对于需要激活两种(或多种)反应物的复杂反应的反应性通常较低。到目前为止,SAC在生产手性构建块中的用途尚未实现,因为其有限且各向同性的配位往往会导致外消旋。这需要对动态催化循环有更深入的了解,以开发用于复杂有机转化的更有效的SAC,这可能涉及双金属SAC的使用。除了催化剂设计之外,SAC在化学生产中的商业化也受到了催化剂制备中缺乏标准协议(即实验室标准之间的差异)以及使用实验室分批反应器的生产力不足(大多在克级或更低)的阻碍。最后,需要开发连续流合成和自动化技术,将实验室实践转化为实际操作中的工艺知识。
在这篇报道中,作者介绍了作者在SAC介导的精细化工生产有机合成以及使用连续流技术扩大该过程方面的贡献。在催化剂水平上,作者将通过操作和定量技术提供对动态催化循环的分子水平理解,以设计更有效和耐浸出的SAC。将特别关注SAC介导的反应机制,该机制可以产生增值药物。在反应堆层面,作者将重点介绍反应堆内部的多尺度物理和实验技术,以进一步提高生产率。最后,将从这个角度讨论SAC研究中需要克服的技术障碍,如生产成本、高通量筛选和新的反应器设计。
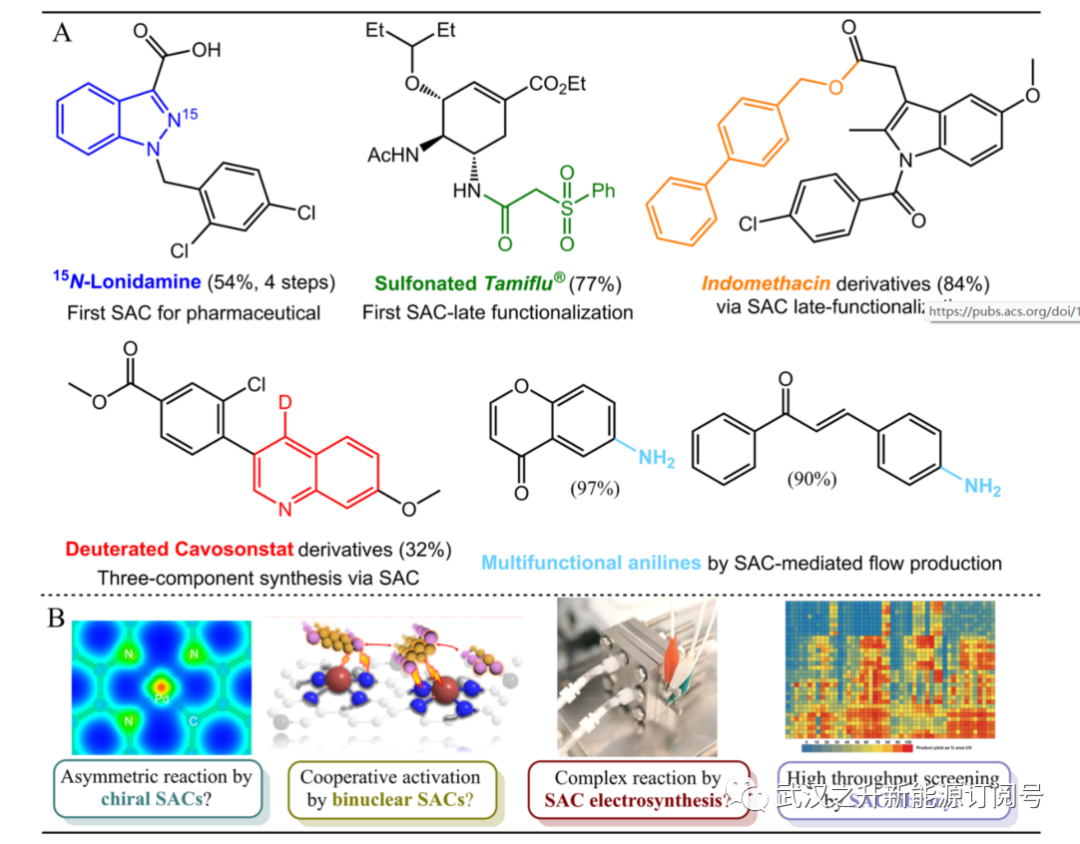
图1. 通过SAC介导的有机合成生产增值化学品。(A) SAC生产的典型药品。转自参考文献7、11、16。
【主要内容】
2迈向更高产、更耐浸出的SACS之路
原子水平对局部配位环境的调节是提高SAC在液相有机反应中催化性能的有力策略,这在几篇开创性的综述中已经得到了充分的证明。尽管如此,由于有机物的复杂性和缺乏解决SAC在此类反应中的作用的能力,这种设计可能是经验的。在下文中,作者将提供作者的观点和全面的画面,从原子设计到微观、中尺度和宏观尺度的工程,以合成更高效、更耐用的SAC。
2.1活性位点的定量鉴定
由于支撑矩阵的复杂性,不同的配位位点可以在同一批次的特定SAC中共存。例如,在典型的缺陷稳定SAC中,金属的配位环境受到矩阵中缺陷类型和分布的影响。这意味着,不考虑底物基质的SAC的简单原子模型通常不能真正代表性,并模糊了对反应如何发生的理解。此外,当存在具有不同配位位点的各种SAC时,具有较低种群的SAC可能是催化热点,而不是大多数SAC。呼吁开发定量技术,将局部配位环境与催化活性联系起来。
不幸的是,原子分辨率扫描透射电子显微镜(STEM)仅在局部区域提供有限的信息,而X射线吸收光谱(XAS)在区分次要物种和大量信息方面不敏感。3良好的实践应采用工具集和统计方法相结合的方法进行定量测定(图2)。例如,通过XAS中的曲线拟合和模拟,定性地获得了Pt在作者的Pt1−MoS2催化剂中的平均配位环境。每个Pt原子与三个S原子共价键合,形成强结合的金字塔结构,催化硝基芳烃化学选择性还原为多功能苯胺(图2A)。STEM的进一步统计分析证实,大多数Pt原子位于Mo顶上位置(171个计数中93%),而一小部分(~7%)原子位于蜂窝结构中。硝基在大多数SAC上的优选吸附通过理论计算得到证实。在另一项工作中,通过STEM定量探索了具有不同Cu负载量的Cu1−C3N4 SAC中的原子间距离,得出21、9和4wt%Cu SAC(样品尺寸分别为410、245和163)的平均距离分别为0.74±0.13、1.58±0.50和2.43±1.02 nm。这导致作者在SAC中通过减少反应物典型扩散长度内的最近邻距离来开发双核型催化。
SAC的定量分布与催化活性的相关性提供了对主要活性物质的更深入了解。对于作者的Fe1 SAC介导的氧化环化,传统的吡啶类Fe−N4物种(D1,亚铁的低自旋)由于其在基质中的丰度(53−72%)而被认为具有催化活性(图2C−E)。然而,通过Fe-穆斯堡尔谱光谱的定量鉴定表明,催化活性与中间自旋物种(D3、Fe2+或Fe3+,S=1或3/2)的百分比密切相关,其中最具活性的Fe-800 SAC也具有最高的D3含量(~19%)。相反,高自旋物种(S=2的Fe2+,D2/D4双态)由于其饱和配位而通常是不活跃的。这与Fe−N−C催化剂选择性氧化C−H键的发现非常一致,其中中间自旋物种的活性比吡啶类Fe−N4物种高约1个数量级。如果没有活性位点的定量测定,识别主要活性SAC是不可能的。作者应该记住,这种鉴定容易产生环境造成的伪影,例如,氧分子吸附在Fe物种上会增加配位数。因此,有必要在操作条件下进行XAS等实验。
2.2动态催化循环的操作研究
SAC可以在催化过程中经历结构重建为催化活性形式(例如在CO2还原过程中由原子分散的铜形成金属簇);它们还可以阻止矩阵的重建以提高长期稳定性。因此,XAS等操作技术可用于直接观察反应过程中键配位的动态变化。在接下来的研究中,作者定制了一个具有双面Al涂层的硬币电池型反应器,以防止有机溶剂和侵蚀性反应物对Kapton窗口的腐蚀,从而使反应持续24小时,这与实验的时间尺度相当。
如图2F所示,Cu1催化的腈−叠氮化物环加成过程中的动态结构演变反映在Cu K-edge X射线吸收近边结构(XANES)光谱中上升沿的肩峰中,这源于Cu−N4构型引起的未占4p轨道能量的分裂。叠氮化钠在Cu-SAC上的初始激活导致蓝移,由于[dp3]方形金字塔构型的形成,肩峰强度降低。同时,叠氮化物和苄腈在Cu中心发生配体交换将在较低的能量下提供更尖锐的肩峰。4小时后,反应顺利进行,形成具有稳定XANES模式的四唑产物。这与动力学研究中的线性区域(5−15小时)一致。16局部配位环境的演变也可以通过图2G中操作傅立叶变换扩展X射线吸收精细结构(FT-EXAFS)的变化来跟踪。Cu对叠氮化物的活化导致Cu−N配位幅度明显增加,而随后的配体交换和双核活化过程将经历净减少,这是由于叠氮化物或腈在四唑形成时在附近的Cu位点上分离。Cu−N配位幅度一直保持在低强度,直到反应完成,表明在Cu1−C3N4催化剂中双核途径占主导地位。Li和Wang等人还开发了另一种用于复杂液相反应的操作XAS反应器。以定量或单分子方式进行操作测量(例如,穆斯堡尔谱或实时成像)将非常有趣,为SAC介导的不对称和手性反应铺平方向(图1B)。
2.3.动态粘合与耐浸出性
SAC的一个主要优点在于它易于从反应介质中分离。然而,SAC在液相有机转化中的作用受到溶剂和配体存在下浸出的阻碍。在填充床反应器中的流动操作下,这种问题变得更加严重,其中需要高的操作压力(通常为10bar及以上)以获得令人满意的流速。SAC的耐浸出性从根本上与金属原子与其配位原子之间的局部结合强度有关。与纳米颗粒催化剂相比,SAC通常更耐金属溶解和表面迁移,因为它们具有更强的结合力。然而,动态催化循环也需要一定程度的结构灵活性。在Pd1−C3N4催化的Suzuki偶联的情况下,Pd原子在跨催化过程中与基质的配位较少(2.6对6)。这种较弱的结合强度增加了金属浸出的可能性;因此,在催化活性和稳定性之间存在权衡。
使用不对称配位(例如,M−N3C1而不是M−N4 SAC)将是解决这一困境的可行途径。在这种情况下,通过安装较弱的键(如M−C)来确保结构灵活性,而金属通过强键(如M−N或M−S)与基体保持强键合。例如,作者从Pd修饰的N-混淆四苯基卟啉开发了一种预定义的Pd−N3C1 SAC,用于Suzuki偶联。与氮配体相比,由于碳的吸电子性能较弱,金属中心的不对称电荷密度促进了氧化加成中间体对苯基硼酸的亲核攻击,并且还降低了随后的跨催化的能垒。所制备的Pd-SAC显示出良好的循环稳定性。热过滤实验证明了Pd−N3C1 SAC的稳定性。同样,Cao等人也开发了一种Co1−N3P1 SAC,其对硝基芳烃的氢化具有前所未有的高活性和选择性。通过STEM中的原子尺度N K边和L边电子能量损失谱(EELS)图验证了Co1中心的不对称配位。密度泛函理论(DFT)进一步揭示了这种不对称构型的−0.864 eV的形成能,证明了与传统的面内Co1−N4 SAC相比较的稳定性。
另一个有趣的协同机制来自相邻SAC原子对反应物的协同活化。为了同时激活多种反应物,SAC必须变形并与基质脱配。很难只有单一的位点机制来催化复杂的有机转化。为了获得多站点机制,一种可能性是让两种附近的金属在超高负载SAC中协同工作。作者最近的研究表明,将金属间距离减少到反应物的典型扩散长度(<1 nm)可以有效地促进双分子腈叠氮化物环加成达到几乎相同的产率(87%分离)。这一点得到了操作XAS测量的证实(图2F,G),并得到了根据扩散驱动碰撞理论反应概率对原子间距离的准线性依赖性的支持。这种概念可以进一步扩展到使用超高负载Cu-SAC氧还原、叠氮化物–炔烃环加成和乙炔氢氯化、和类芬顿反应的无钯Wacker氧化。最重要的是,在动态流动条件下操作需要SAC和基质之间的牢固结合。为了解决这个问题,作者制造了一种具有棱锥状Pt-3S配位的Pt1−MoS2催化剂,以避免在流动反应器中长期操作过程中金属浸出。关于流动操作整体式催化剂的详细信息将在下一节中介绍。
2.4用于生产操作的整体式催化剂
SAC研究中长期缺失的部分在于将实验室粉末基催化剂转化为工业上可行的催化剂配方。这种技术催化剂通常在组成、结构和孔隙率方面与粉末催化剂有很大不同,以表现出工业上兼容的物理(质量/传热)、化学(功能),或机械(强度和耐磨性)特性。在这方面,需要更全面地了解单片SAC在连续流生产中的微观、中微观和宏观结构。
如图3A−C所示,作者使用天然丰富的木材材料作为支架,制造了机械强度高、分级多孔的SAC。在木材加工过程中,通过简单地加入金属离子前体(FeCl3、CoCl2、NiCl2或CuCl2),在与外部氮源以>20 g的批量生产率热解后,可以获得具有M−N4配位的稳定SAC。与使用分子前体的自下而上的策略相比,这允许更节能和更简单的工艺来创建通道和制造整体催化剂。扫描电子显微镜(SEM)证实了在热解后反应物的快速质量扩散动力学中,在平行和垂直方向上保持了各向异性的3D结构。这种整体式催化剂具有机械强度,可以在不变形的情况下支撑其自身重量的650倍以上。同样,氧化还原液流电池中的市售石墨毡(GF)也可用作催化剂载体。GF涂有水热生长的MoS2,用于随后固定单个金属原子,包括Pt、Fe、Co、Ni和Cu。如图3F所示,这种单片SAC表现出比纯GF高得多的抗压强度,并且能够耐受至少五次连续测试,具有90%的形状变形,而不会发生结构退化或粉末脱落。此外,如水接触角测量所证实的,这种催化剂的表面对于流动操作是高度可润湿的。
对于互连的大孔结构在解决从本体溶液到催化剂表面的外部传质限制方面的作用,人们没有给予足够的关注。在大多数SAC报告中,进行了Brunauer、Emmett和Teller(BET)分析,以提供关于中孔/微孔结构的信息,用于评估表面积,这与粉末样品中纳米颗粒之间的内部传质有关。然而,对于反应物的定量转化,外部质量扩散传输在低流速下主导催化剂性能,需要优化大孔结构以提高催化活性和稳定性。作者已经应用X射线显微成像(micro-CT)、压汞孔隙率测试、SEM和计算方法来表征单片SAC中的互连通道网络。与BET分析不同,压汞孔隙率测定法提供了关于溶液可接近的大孔结构的有价值的信息。在图3E中的Co1−MoS2 GF中观察到非常大的孔,其典型孔径为~65μm,孔隙率为91%−95%,这有利于在中等压力下的快速扩散动力学。溶液可接近的比表面积可以通过使用甲基蓝和/或其他分子的吸收测试来评估。通过微CT分析,可以从真实催化剂中重建具有高空间分辨率~100 nm的真正具有代表性的3D模型,显示Co1−MoS2 GF催化剂中错综复杂的互连通道网络(图3D)。在压缩下,孔隙进一步收缩,催化剂表面可能产生局部湍流。这种3D模型允许通过第4.2节中的CFD计算,详细分析微观结构(如孔隙率、表面粗糙度、收缩、弯曲度)与操作条件下的流动行为之间的联系。
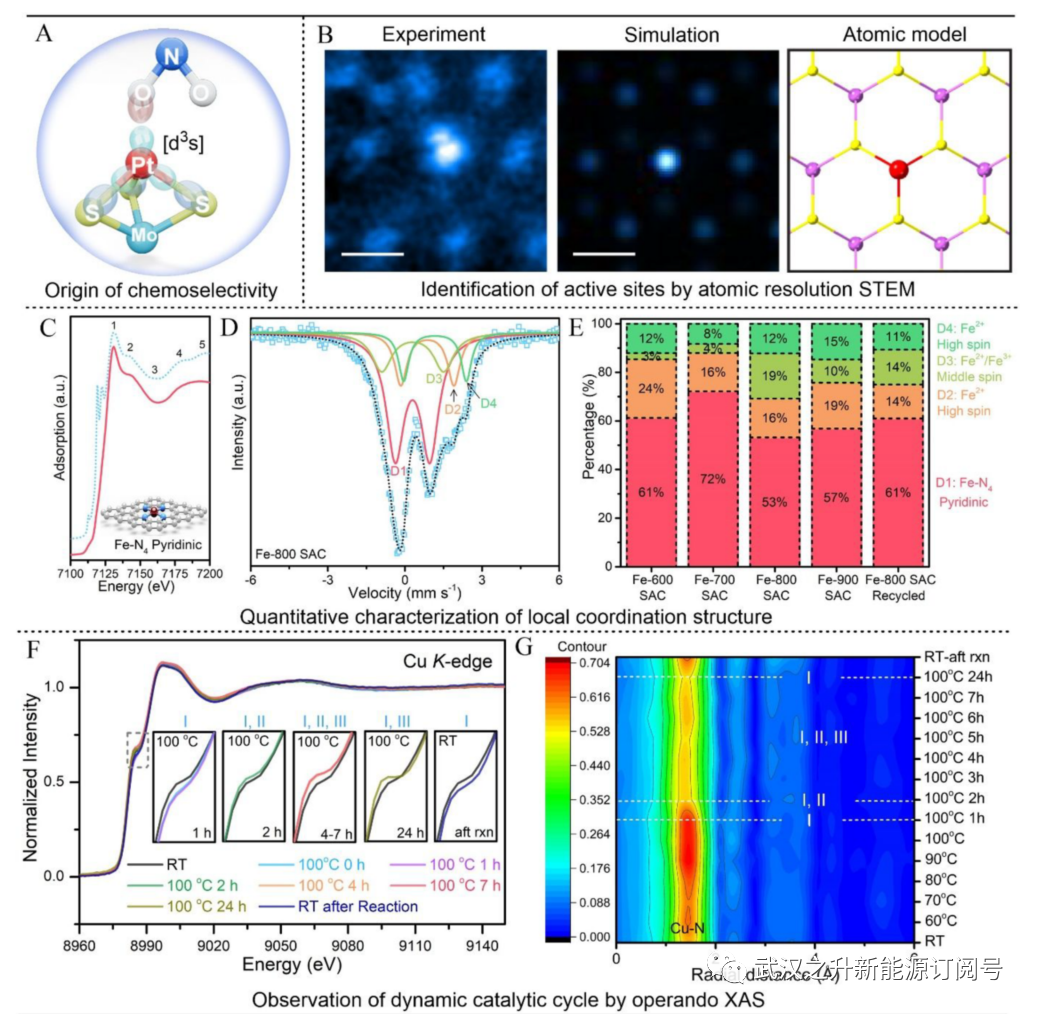
图2. 探索SAC中活性位点的动态催化循环。(A) 硝基芳烃化学选择性还原的Pt-3S金字塔构型示意图和(B)Pt1−MoS2的相应原子分辨率STEM-HAADF图像、图像模拟和原子模型。比例尺:0.2纳米。(C) 实验和计算了具有吡啶型Fe−N4结构的Fe-800 SAC的Fe K边XANES光谱。(D,E)富含57Fe的Fe-800 SAC的室温57Fe-Moåssbauer谱和活性物质的定量分析。(F) 21wt%Cu1−C3N4 SAC的Cu K边XANES光谱及其在腈−叠氮化物环加成过程中的演变。插图显示了上升边缘肩部峰的演变。(G)21 wt%Cu1−C3N4 SAC在腈−叠氮化物环加成过程中的FT-EXAFS光谱。经(A,B)参考文献15, 9,16许可转载。
3增值药品的反应设计
化学选择性是SAC在液相转化中相对于传统纳米粒子催化剂的固有优势。作者之前已经强调了控制选择性的潜在因素,重点是热力学和动力学方面的考虑,以及在后期通过SAC或通过顺序或多组分反应构建药学和特种化学品。在下文中,作者将简要回顾化学选择性的起源,并强调作者对SAC介导的精细化学产物的新反应设计。
3.1.SAC化学选择性的起源
如图4A所示,与传统多相催化剂上的不同结合位点相比,SAC的化学选择性源于其受限的吸附构型;2,3这已经通过红外光谱、拉曼光谱和DRIFTS等操作光谱研究得到了证明。例如,在原位DRIFTS测量中,与Pt纳米颗粒上烯烃基团(如乙炔(三齿)或二σ(二齿)构型)的平面结合构型相比,作者的Pt1−MoS2更倾向于3-亚硝基苯乙烯中硝基官能团的单齿“端接”吸附。这导致了对3-氨基苯乙烯99%的非凡化学选择性。DFT计算揭示了Pt1−MoS2中沿z轴的高度定向电荷分布,有利于极性官能团的吸附。Pt2+金属中心(d8s0)与附近的三个S原子形成棱锥形[d3s]杂化,为具有缺电硝基官能团的Pt杂化5d6s-O2p键留下沿z轴的半填充轨道。类似地,作者的Co1-in-MoS2催化剂还显示,与其他竞争位点(如硼酸基团中的OH位点)相比,硫位点上垂直构型的硫化物吸附更强,这是对硫化物氧化具有优异化学选择性的基础。8这允许可控的,图4B中的逐步自由基途径,在化学计量量的H2O2下产生半/完全氧化产物(亚砜/砜)。作为合成实用性的证明,使用作者的Co1-in-MoS2 SAC,含有易被传统方法氧化的缺电子烯烃的达菲可以成功转化为亚砜和含砜的类似物(图1A)。
3.2.多位点途径与单位点途径
SAC的化学正交性(独立化学选择性的一个子集)使复杂分子合成的分子催化得以复制。传统上,这是通过顺序、多米诺骨牌或多组分反应的单位点机制进行的,以快速引入分子复杂性。例如,作者报道了Pt1/CeO2促进α-重氮酯的E-选择性氢化,以合成具有生物活性的1H-吲唑及其15N标记的类似物,包括抗癌的山莨菪碱、佐剂、格拉司琼和去甲咪唑的构建块以及生物活性生物碱尼戈林和尼戈林(图1A)。在Pd1−N3C1 SAC存在下,通过顺序酯化和Suzuki偶联,美国食品药品监督管理局批准的抗炎药多功能吲哚美辛可以以84%的产率成功转化为其长链芳香酯衍生物。同时,多组分反应代表了建立分子支架复杂性的另一种有效策略。作者已经开发了一种三组分氧化环化反应,以使用富含地球的过渡金属基SAC(如Fe1、Co1、Ni1和Cu1),以有效、原子经济的方式从原料苯胺和乙酰甲酮中提供各种喹啉(23个实例)。通过一步Fe1催化的环化(35%的产率)而不是传统的Pd催化的Suzuki偶联(五步46%),成功地合成了Cavosenstat衍生物及其4-氘化类似物(F508del CFTR突变的处理)。在优化的条件下,通过Ru3O2/rGO催化的Friedlaånder合成获得了更高的分离产率(47%)。同样,Chen等人开发了一种磷配位的Rh1-SAC,用于使用合成气(H2和CO)进行烯烃的加氢甲酰化,该合成气具有高转化率(>99%)和对支链醛的高区域选择性(>90%,或支链与线性之比为13.3)。这使得布洛芬和芬地林能够以高产率分步和原子经济的方式合成。Wang等人还报道了使用Cu1−TiC SAC对炔烃(直链到支链>100:1)的高度线性E型区域选择性转化,这首次适用于使用硼氢化途径全合成西那卡司盐酸盐。最近,同一小组报告了使用非均相铱单原子位点催化剂的高度区域选择性碳类O−H键插入。使用Co1−1可以在百万分之一的催化剂负载下实现酰基氯的氧化氟化n4@ncSAC.多相有机氮化学合成代表了增值化学品的另一种重要转化,推动了SAC的发展,以快速构建N-杂环。
尽管如此,这种单位点途径在平衡催化活性和稳定性方面可能存在问题(第2.3节)。通过附近SAC原子上的两个(或多个)协同活化,多位点途径产生了一种新的解决方案。如图4C−E所示,作者选择了腈−叠氮化物环加成体系来证明双核介导的复杂有机反应。正如预期的那样,低负载SAC(<9wt%)由于缓慢的单位点途径而受到较差的活性(~10%的分离产率)的影响。相反,21wt%的Cu1−C3N4 SAC,Cu间距离为0.74±0.13 nm,为四唑产物提供了87%的分离产率。这甚至比市售的10wt%Cu/C催化剂(58%)更好。鉴于它们相似的多位点途径,Cu/C的较差性能可能归因于与超高负载SAC相比,Cu颗粒中的原子利用效率较低。此外,由于Cu1−C3N4 SAC中更强的Cu−N键合,可以避免Cu颗粒中的金属浸出,从而在循环过程中提供更好的可重复使用性。合作途径反映在图4E中的动力学曲线中,其中叠氮化物的初始活化以及叠氮化物和腈在附近铜原子上的配体交换导致2小时的诱导期。随后在5−15 h内形成四唑化合物,如第2.2.16节XAS操作研究中所述。最近一份关于有机污染物(双酚a)的类Fenton氧化的报告进一步证实了多位点途径在SAC中的适用性。19由于双位点的形成,Cu1/NG中的Cu−Cu相互作用显著影响反应性在5−6Å的距离处的吸附构型。这显著增强了界面电荷转移,从而促进了转化。
这种多位点途径并不总是有利于有机转化,正如单位点SAC和多位点纳米颗粒之间催化活性的比较所反映的那样。对于Pd1Au催化的Ullmann反应,当大多数金属中心在空间上分离时,在低金属含量下会观察到催化转换的急剧增加。在两种途径中反应性相似的情况下(例如,Pt1/FeOx催化的亚硝基苯乙烯加氢),活性将随着分散度的增加而逐渐上升,直到随着金属物种倾向于SAC而达到平稳。这一现象仍然存在于超高负载SAC中。例如,尽管使用了类似的SAC(分别高达16.3wt%的Ni1/NC、21.8wt%的Cu1/PCN和13.7wt%的Pt1/NC),但对电催化CO2还原、叠氮化物–炔烃环加成和乙炔氢氯化的负载(相互作用)依赖性显著不同。这需要对SAC进行更详细的机制研究,特别是对附近金属的真实功能(例如,电子耦合或扩散碰撞)。另一个有趣的主题是通过具有远程功能的金属连接对复杂底物进行远程活化(例如使用铁催化剂在通常与常见功能单元相邻(β)的反应性较低的位置进行位点选择性烯烃硼化),这在SAC中未实现。
3.3.通过(光)电合成实现的新化学
利用光加速化学反应无疑是可持续化学操作的最有前途的策略之一,可以实现许多有用的转化,如重排、环加成、环化和自由基链式反应,以提供其他方法通常无法获得的复杂分子。特别地,[2+2]环加成提供了从烯烃获得环丁烷衍生物的直接方法。王等人的开创性研究证明了使用多相聚合氮化碳(PCN)催化剂通过光催化[2+2]环加成连续流动生产环丁烷(例如,以g为单位的马格纳沙林)。使用市售的玻璃纤维和玻璃珠作为碳腈光催化剂的半透明载体。玻璃纤维上的有效催化剂负载量约为1.2 wt%,可以通过简单地涂上饱和尿素溶液,然后在马弗炉中热解将尿素转化为PCN来实现。考虑到PCN是固定SAC的一种流行的富氮载体,特别是对于高达21.8wt%的超高负载SAC,这种光化学流动技术可以很容易地用于SAC介导的精细化学品生产。目前,这是一个开发较少的研究领域,使用SAC的光催化转化仅限于光催化水分解、CO2还原和氧化分解。
SAC介导的(光)电合成可以串联操作,以快速构建复杂分子。Wu等人报道了一种独特的卵黄壳Pd1@Fe1由氮掺杂碳壳中原子分散的Fe1位点和衍生自金属有机框架的蛋黄中的Pd1位点组成的催化剂。蛋黄壳Pd1@Fe1允许直接利用电化学水分解中的氧和氢,同时催化硝基芳族和二苯基二硫醚的氢化和烯烃环氧化反应,以级联方式分别产生7种氨基醇(72−91%产率)和6种羟基硫化物(58−82%产率)。在超过13小时的连续回收测试中观察到具有高生产率和高选择性的稳定性能。这些发现提供了一种将不同的单一金属位点整合在一个系统内的通用策略,以允许连续且容易地合成用于各种具有挑战性的反应的复杂化合物。
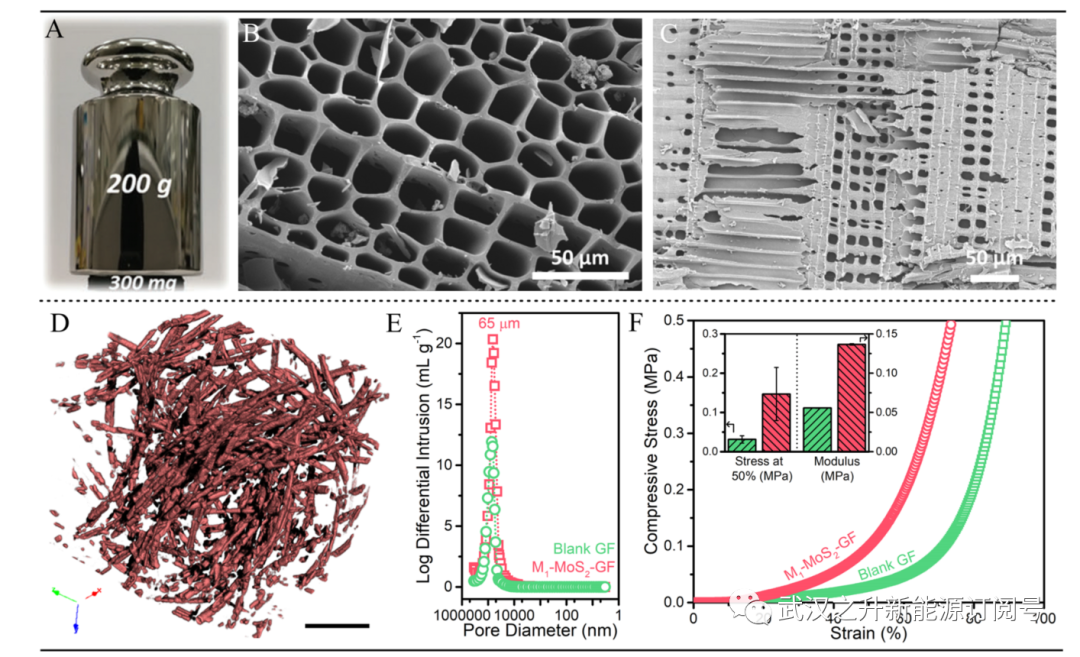
图3.单片SAC催化剂的制造。(A) 支撑667倍重量的整体式催化剂(300mg)的图像。(B) 碳化后在平行和(C)垂直方向上的SEM图像。(D) Co1−MoS2 GF的假彩色显微CT图像。(E)压汞孔隙率测定法和(F)空白GF和Co1−MoS2 GF的压缩应变–应力曲线。插图显示了相应的应力和压缩模量。比例尺:(B、C)50μm;(D) 100μm。经(参考文献38,15许可复制。
4.对提高生产力的反应性理解
连续流化学是制药工业中使用的一种通用方案,通过促进多相界面的质量扩散动力学并最大限度地减少催化剂分离过程中的废物产生来优化反应。一个主要原因在于流路的缩小,这显著提高了多相反应的相混合效率。流动反应器还允许在恒定浓度下形成产物,这对于最大限度地减少由于过度反应造成的选择性损失至关重要。各种在线分析和在线分析的结合实现了快速反应筛选,以实现流程优化。典型的连续流装置可能包含八个基本单元:流体和试剂输送、混合、反应器、骤冷、压力调节、收集、分析和纯化。在下一节中,作者将介绍如何将快流反应器设计用于SAC介导的液相有机转化。
4.1.用于有机转化的快流反应器
快流反应器是一种具有平行板和/或膜的流动系统,以堆叠方式组装,并已用于液流电池、燃料电池、CO2电解槽和许多其他电化学应用中。与催化剂粉末密集填充到催化剂柱中的填充床反应器不同,快流反应器通常使用气体扩散载体(如碳纸)来安装催化剂(出于导电目的),这允许反应物在开放通道中的高流速(液体>10 mL min−1)。通常向快流池施加外部电压,以促进电极上的反应动力学,从而补偿高流速下减少的停留时间(通常τ<5分钟)。尽管许多报告已经证明了在SAC催化的电化学反应中使用快流反应器(如氧气和二氧化碳还原),精细化学品的生产流程仍然是一个挑战,因为与填充床反应器相比,它们的转化率不令人满意。
最近,作者开发了一种SAC改进的快速流动原型,用于以前所未有的生产力定量生产精细化学品。选择硫化物的化学选择性氧化作为模型反应。与之前的分批操作中的20分钟相比,流动操作中的反应可以在30秒内完成。短的反应时间对于最大限度地减少后期功能化过程中不期望的副反应是至关重要的。完全氧化产物(甲基苯基砜)的生产率可以在10倍浓度(0.25 M硫苯甲醚)下进一步提高到4.7 g h−1,而不会降低转化率和选择性。硝基芳烃的化学选择性还原进一步验证了这种实用性,这是生产多功能苯胺的工业上重要的反应。如图5所示,作者的SAC液流电池配备了Pt1−MoS2催化剂模块(3.2 mg Pt),具有极高的化学选择性(>99%)和生产率(苯胺>5.8 g h−1),用于生产昂贵的构建块,包括28种多功能苯胺,如6-氨基-4H-色烯-4-酮(约300美元g−1)。与实验室分批工艺相比,液流电池的生产率显著提高是一个主要优势(对于相同的反应,0.02−0.07 g h−1)。值得注意的是,3-氨基苯乙烯,一种重要的原料化学品,可以通过使用Pt1−MoS2液流电池以99%的产率还原3-硝基苯乙烯来有效生产,这与使用Pt/C控制的<10%的产率形成鲜明对比。作者的连续流方案也适用于蒽和许多杂环,包括吡啶、喹啉、羟吲哚和邻苯二甲酸酯。由于金字塔状Pt-3S结构的稳定性,所制备的Pt1−MoS2催化剂具有高度的抗金属浸出性,允许流动反应器在高流速和各种转化率下长期运行,而不会降低性能。这一点通过0.1至20 mL min−1的流速测试得到了证实(图5C),其中可以实现连续24小时运行的高度稳定性能。
与电化学转化相结合的能力是快流反应器的一个巨大优势。通过提供电子和电子空穴形式的“无痕迹”试剂,电化学合成越来越多地被合成界所采用,以避免化学计量的氧化剂和还原剂,与传统方法相比,具有优异的化学选择性。已经致力于SAC介导的流动电合成。例如,在氮掺杂碳上使用铁SAC的快流池中,在50小时内以215μmol cm−2 h−1的前所未有的生产率将一氧化氮选择性还原为羟胺。然而,由于有机转化的复杂性,它尚未扩展到有机电合成。
4.2.流动反应器内的多尺度物理
在SAC的研究中,反应器内多尺度物理的影响一直被忽视,因为大多数工作都集中在原子水平的材料工程上,以微调催化剂和反应物之间的相互作用。由于流动反应器的复杂性、质量和传热动力学、孔隙率和曲折度的局部变化,和催化活性表面积(即润湿性和气–液–固界面)对性能有着深远的影响。当反应由传质动力学控制时,这些性质特别重要。在这种情况下,来自催化剂固有活性的影响变得不那么突出,因为瓶颈是由反应器水平的工艺参数和工程决定的。
应通过低转化率(<30%)下的流速测试仔细检查质量扩散限制的程度。如图5A所示,在中低流速下,外部质量扩散限制主导了作者的液流电池性能,而在7.5 mL min−1或以上的流速下,它会受到反应限制。这表明流动反应器中的催化剂筛选应在扩散和反应受限的条件下进行(图5B),以便在SAC和对照催化剂之间进行公平的比较。同时,应在高流速下验证金属浸出(稳定性)和化学选择性,以排除质量扩散限制的影响。例如,作者的Pt1−MoS2 SAC在高流速和低转化率(<30%)下对4-硝基苯乙酮的4-氨基苯乙酮表现出>99%的化学选择性,证实了其来源于SAC上反应物的有利化学吸附。
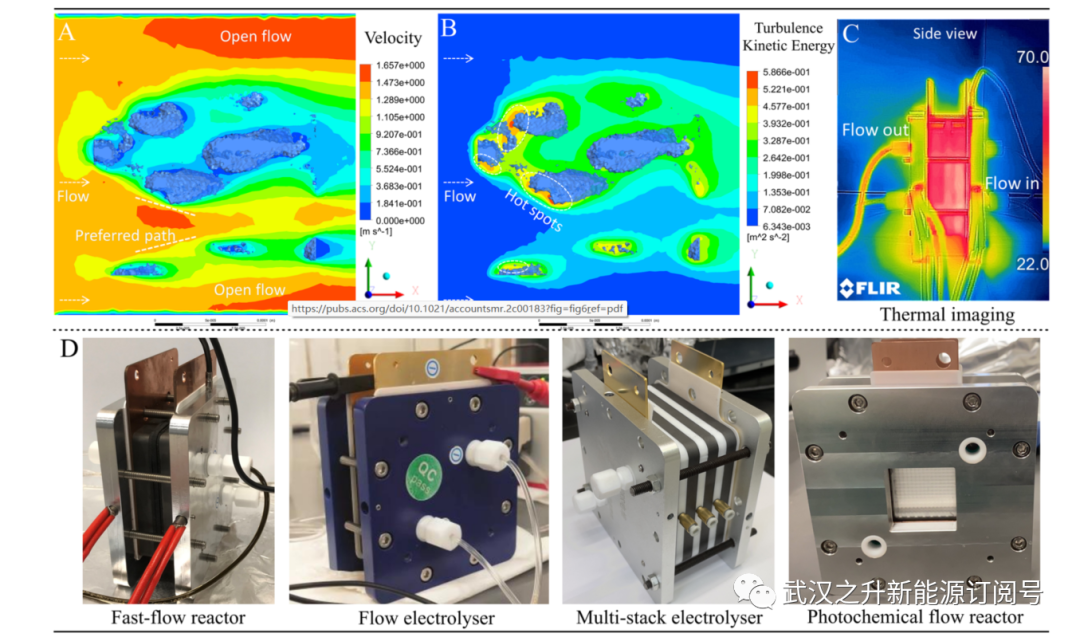
除了动力学分析,CFD计算是可视化反应器内流体行为的强大工具。如图6A,B所示,作者已经确定在碳纤维之间的狭窄空隙空间中存在流体热点和优选的流动路径,由于在催化剂表面附近促进了传质动力学,这与催化转化率具有很强的相关性。通过压缩催化剂模块来调节流体热点会在流动路径中产生更强的局部湍流,并在扩散受限的情况下显著提高反应器性能50%以上,这在SAC研究中从未得到证实。直接观察反应器内的速度(和流体分布)可以使用带有X射线透明聚醚酮(PEEK)盖板的操作式液流电池或带有石英窗的热成像来实现。51从氧化还原流动电池中得到的启示来看,通常有利于窄流动通道以促进传质动力学。此外,在操作条件下,通过热像仪对反应器内的热分布进行成像(图6C),显示了催化部件处的均匀热分布和由于有效的热交换而产生的热流出溶液。流出溶液的温度迅速下降到环境温度。与分批操作相比,在流动反应器中以快速加热和冷却轮廓对敏感功能的这种处理对于最小化不希望的副反应是有利的
优化气–液–固界面是SAC介导的电催化的另一个重要方面,它也与液相有机转化有关。在电化学CO2还原中,水驱是快流电池中的一个常见问题,它通过阻止气体扩散到催化剂表面而显著降低反应器性能。同时,脱水过程降低了法拉第效率并降低了CO2的利用率(能耗)。因此,对催化剂表面润湿性的仔细控制对于多相界面系统至关重要。气–液(或液–液)混合物中的有效相混合对于生产反应性也是必要的。增加界面表面积是将传质(混合)提高2个数量级的有效方法。因此,使用较小的颗粒或3D分级支架来增强反应动力学是有益的。54催化剂表面(通道壁)上的气液混合物中可能出现三种典型的流动行为,即气泡、段塞和环形流。这些行为受到流速、粘度和表面润湿性的影响;因此,应该对工艺参数(如电解槽中的气体/液体流量)进行全面的研究,以优化液流电池的性能。
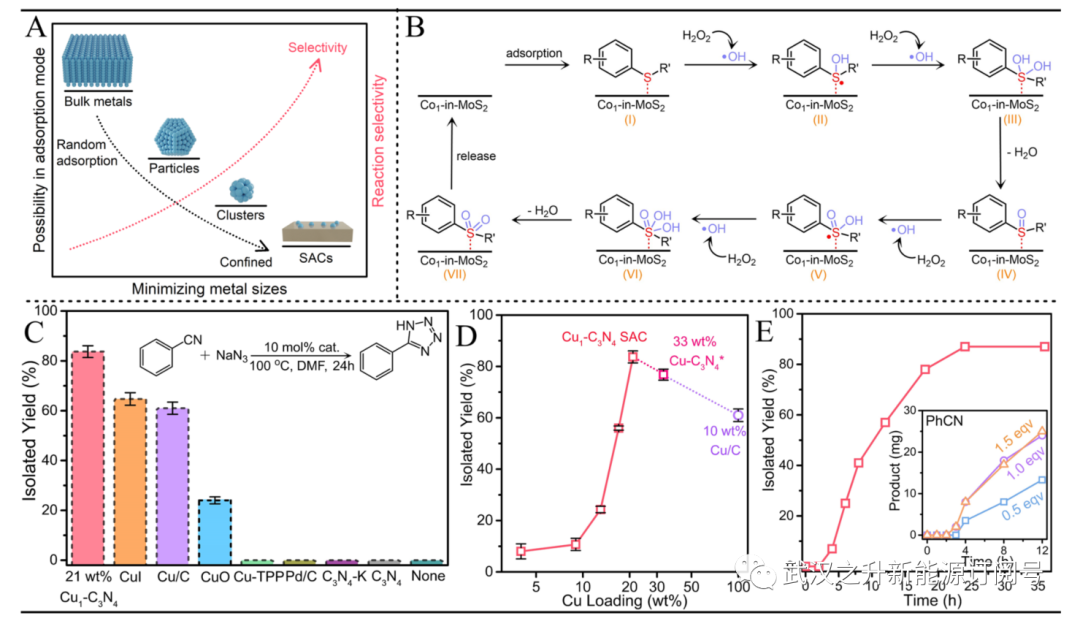
图4. 精细化学品SAC生产的反应设计。(A) 反应物的可能吸附构型和每个金属原子在传统支撑基底上的反应选择性与金属尺寸的关系。(B) Co1−MoS2催化硫化物氧化的自由基介导途径;。(C) 腈−叠氮化物环加成的催化剂筛选。(D) 在相同的Cu用量(10 mol%)下,Cu1−C3N4催化剂的催化性能对Cu负载量的依赖性。(E) 在标准条件下监测21 wt%Cu1−C3N4随时间变化的反应。插图显示了动力学研究。经参考文献3,8,16许可改编。
5对SAC介导精细化工生产的展望
新催化剂的商业化是一项有风险的事业,需要克服技术和经济障碍。在下文中,作者将讨论这些障碍以及未来通过SAC生产高价值化学品的可能解决方案。
5.1 SAC的标准化和可扩展生产协议
缺乏标准化和可扩展的生产协议是SAC研究的一个主要瓶颈。大多数报道的SAC催化反应只产生克级产物,迄今为止还没有扩大批量工艺的报道。另一个问题是活性原子迁移和聚集到活性较低(选择性)的纳米颗粒中通常发生在反应过程中,导致产物的再现性较差。球磨(或机械化学合成)、浸渍、和通过煅烧的热稳定SAC代表了SAC生产的三种主要方法。SAC的公斤级批量生产已得到证明(表1)。就SAC制备而言,球磨技术的一个缺点在于单个原子聚集成更大的纳米颗粒。这种不稳定性可以通过引入强的金属–衬底相互作用来解决,从而通过简单的煅烧批量生产热稳定的SAC。例如,Pt1/Fe2O3催化剂可以通过在800°C的空气中煅烧负载在Fe2O3上的Pt纳米颗粒,形成稳定的Pt−O−Fe键来获得。同样,Li等人报道了通过在N2中在900°C下加热将贵金属纳米颗粒直接转化为热稳定的单原子。然而,这种策略通常仅限于富含配位原子的可还原氧化物或载体。浸渍可以说是SAC工业制备最有用的方法,特别是实现有机转化的超高金属负载。值得注意的是,Lu等人开发了一种两步退火方法,用于控制配体的去除,以防止未结合的金属前体在催化剂载体上的热诱导聚集。该方法允许在氮掺杂的碳、聚合氮化碳和氧化铈上制备用于超过15种过渡金属的单金属或多金属超高密度SAC(高达23wt%),并且可扩展到千克水平(1.88kg,20.8wt%Ni)。该协议的自动化实现了对SAC合成过程的精确控制(例如,金属添加的数量和速度、样品洗涤)。
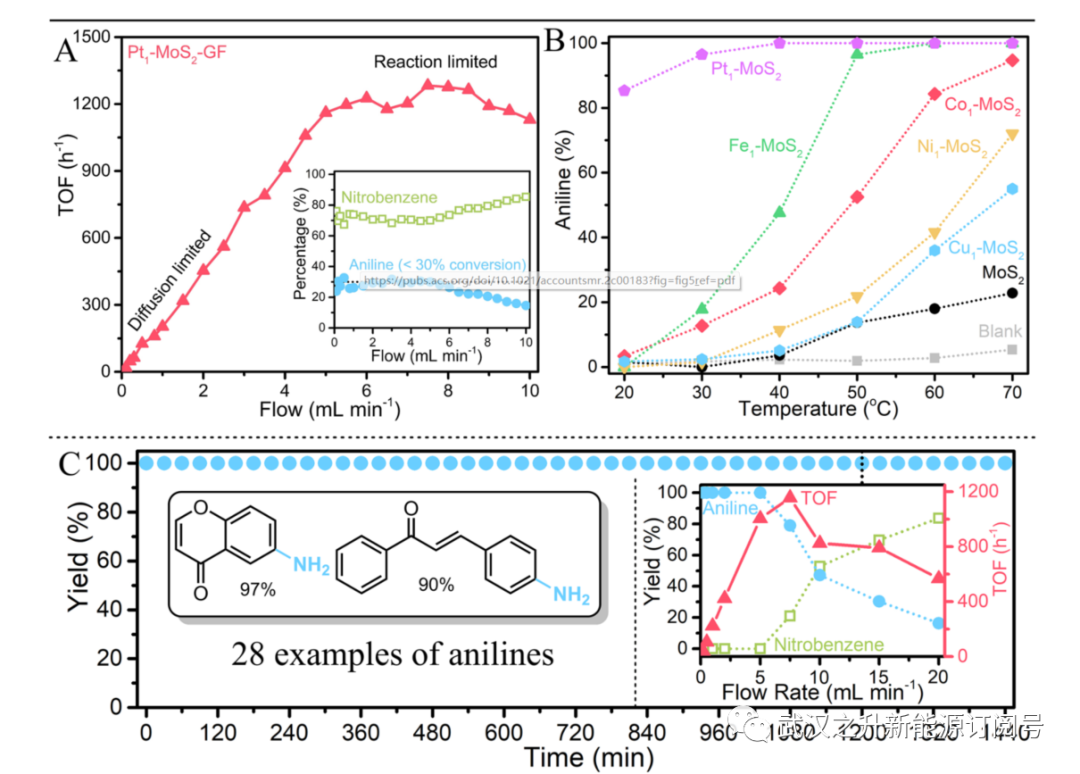
图5. 液流池性能。(A)TOF与流速的关系图,显示了液流电池性能中的质量限制状态和反应限制状态。插图显示了在不同流速下反应物和产物的百分比。(B) 各种SAC的催化剂筛选和定量转化体系中的对照。(C)Pt1−MoS2 GF催化剂在定量转化制度下的24小时运行演示。插图显示了通过流动反应器生产的多功能苯胺的化学结构(多达28个实例),以及在第20小时的流速测量。在参考文献15中可以找到反应受限状态下的液流池性能。
5.2 SAC催化剂的成本评价
催化剂的生产成本是任何扩大规模过程的重要经济考虑因素。由于缺乏公共领域的信息,特别是在工业规模合成催化剂的资本和运营成本方面,催化剂的成本评估通常对实验室团队来说是具有挑战性的。根据千克生产的现有协议,作者采用了一种简化的分步法(CatCost,美国能源部1.1.0版),以使用球磨和湿法浸渍法估算四种典型SAC的吨级生产成本。在这种方法中,从已建立的数据库中选择与催化剂合同制造商使用的特定工艺设备相对应的单个加工步骤,该数据库列出了原材料的价格、用于大批量合成的专用催化剂生产厂、废催化剂价值以及其他重要特征。
如表1所示,球磨方法显示出工业规模生产的最佳成本效益。按照每天1吨的生产规模,1.1 wt%Fe−N−C SAC的生产成本估计为每公斤20.44美元。该成本远低于商业负载的催化剂,例如2重量%Pt/C(每公斤75.15美元)和21重量%Ni/Al2O3(每公斤47.02美元)。与热稳定SAC的湿法浸渍和煅烧相比,球磨的主要优点是减少了操作时间和工艺成本。值得注意的是,当生产Pd1/ZnO催化剂的球磨时间缩短到10小时时,工艺成本可以降低到每公斤约2.5美元。在这种情况下,原材料(贵金属盐)的成本占总生产成本的96%。因此,对于贵金属SAC,有必要从废催化剂中回收金属以降低生产成本。随着回收,1.8wt%Pt1/FeOx-C800SAC的价格与商业2wt%Pt/C兼容(84.23对75.15美元/公斤),尽管初始成本可能高出约6倍。同时,应避免使用多个煅烧步骤,因为这会增加工艺成本。在20.8wt%Ni1/PCN催化剂的情况下,工艺成本(每公斤30.18美元)是材料成本的两倍,这增加了规模扩大的复杂性和不确定性。根据粗略估计,超高负载SAC的生产成本对于商业化来说并不高(每公斤71.76美元)。

表1 用既定步骤法对SAC制备的成本评估
5.3 高通量筛选平台
由于金属、载体和化学计量的组合几乎是无限的,使用自动制备系统可以快速生成SAC库来研究潜在的催化反应。37传统上,催化剂评估是按一个接一个的顺序进行的,导致效率和一致性较差。开发一种用于并行分析大量SAC的高通量流量系统是非常可取的。2015年,默克公司展示了对小型自动化平台的开创性研究,在一次实验运行中结合多次注射使用高效液相色谱–质谱法进行分析,以快速无标记地测定与不同产品的反应。通过在室温下在不到一天的时间内使用0.02mg的材料,其中成功的反应实例被进一步转化为克级合成。辉瑞公司的一个研究团队使用单个模块化单元开发了一个自动流动合成平台,用于纳米摩尔规模的快速筛选和微摩尔规模的合成;模块化单元可以以每天>1500个反应的速度进行5760个反应(例如,如图1B所示的热图)。这允许在50−200 mg的范围内确定传统流动和分批模式下复制的最佳条件。最近,Cooper等人开发了一种移动机器人系统,用于筛选水制氢的光催化剂。在8天内用10个实验变量进行了688次实验后,移动机器人确定了一种光催化系统,与最初的配方相比,该系统的活性是原来的6倍。63高通量系统与新型集成人工智能(AI)算法的集成非常有希望探索新的化学物质。64例如,利用基本上所有已知有机化学的训练数据集,通过深度神经网络和符号人工智能实现了对最有前景的逆转录合成路线的自动搜索。这些来自现有平台的知识可以转化为SAC领域。
5.4用于(光)电合成的新型流动反应器
如第3.3节所述,通过光和电合成实现的新化学在增强SAC对复杂有机转化的催化活性方面非常有希望。在快速流动条件下的操作允许更有效和均匀地照射光化学反应混合物,并降低电化学溶液电阻,从而有助于以可重复的方式进一步扩大到制备量。然而,用于析氢或CO2还原的传统流动电解槽不适合SAC介导的有机转化,因为它们不耐受腐蚀性有机溶剂,并且体积太小,无法进行定量转化。通常,有机电合成中报告的电流密度比水电解中使用的电流密度低至少一到两个量级,这是因为溶液电阻高得多,反应物浓度因溶解性差而稀释,并且在复杂的反应路径中存在副反应。这要求高负载,体积大的催化剂电极,具有足够长的停留时间以增加定量转化率,不包括传统的薄膜电极和气体扩散碳纸电极。如图6D所示,作者为SAC介导的光合成和电合成定制了几种类型的流动反应器。为了最大限度地减少有机溶剂的腐蚀,16cm2的单粘性液流电池配备有不锈钢盖板、镀金集电器、具有平行流动路径的石墨板和防腐含氟聚合物管(如全氟烷氧基烷烃(PFA)和聚四氟乙烯(PTFE))。初步结果显示,在液流电池中使用作者的Pt1−MoS2 SAC几乎完全转化硝基苯,在各种电压下对偶氮苯、偶氮苯或苯胺具有高选择性。通过耦合苯胺的氧化和硝基芳烃的还原而进行的成对电解进一步实现了偶氮苯衍生物的克级生产。对于涉及反应的气体,这样的电解槽可以扩展到50 cm2的多叠层形式。石英窗可以安装在这种液流电池的盖板上,用于光(电)化学中的应用。
最后,必须考虑分离膜对反应器性能的影响。聚合物膜(Nafion等)在有机溶剂中的溶胀会导致产物从阴极室到阳极室的交叉(反之亦然),这会显著降低液流电池在转化率和选择性方面的性能。固体分离器(如陶瓷膜)的使用非常有希望长期运行。此外,基于粉末的SAC可以集成到单片膜反应器中,以提高生产率。一个很好的例子是在CO2电解槽中用膜电极组件(MEA)代替气体扩散电极(GDE),这消除了阴极电解液主流并降低了欧姆电阻。然而,催化剂和膜之间的紧密接触可能导致界面处的显著pH和浓度梯度。由于相分离(如无机盐的沉淀),这可能会改变产品分布并降低长期稳定性。因此,要充分发挥SAC催化剂的潜力,需要对流动化学和反应器配置进行系统研究。
上述图片所展示的流动反应器由武汉之升新能源有限公司提供
图6. 了解反应堆水平以提高生产率。(A)基于微CT的真实催化剂模型流体行为的速度和(B)湍流动能的CFD计算。流体热点和首选路径由开放的白色循环和箭头突出显示。(C) 在70°C下运行的液流电池的热成像(侧视图)。(D) 作者实验室中的各种流动反应器用于热、电化学和光化学转化。(A−C)经参考文献15许可复制。
【主要结论】
SAC是生产精细化学品和特种化学品的均相催化剂的一种很有前途的替代品。高的固有催化活性是催化剂成功的一个因素,但SAC的商业化取决于扩大规模的经济性和反应器效率。在这篇文章中,作者回顾了高选择性和耐浸出SAC的设计原理,并讨论了如何通过调节金属间距离来获得多途径。作者还讨论了如何优化反应器内的质量和热量传输。强调了使用SAC催化的流动化学在5.8 g h−1下连续生产多功能苯胺和在4.7 g h−l下连续生产砜,与实验室分批工艺相比,生产率提高了近100倍。对催化剂制备的标准化和经济规模化进行了探索。高通量筛选还有很大的空间来加速新反应的发现。
【文章引用】
Zhongxin Chen, Jia Liu, and Kian Ping Loh, Engineering Single Atom Catalysts for Flow Production: From Catalyst Design to Reactor Understandings.2022,ACS Partner Journal Acctounts of materials research.