
第一作者:黄志峰
通讯作者:黄志峰
通讯单位:湘潭大学
DOI:10.1016/j.ensm.2026.105346

本研究报道了一种通过竞争性吸附诱导的界面重构机制,有效解决了界面稳定性与反应动力学之间长期存在的权衡问题。通过引入羟丙基-β-环糊精(HP- β-CD)和氯离子阴离子,形成了动态非配位相互作用:HP-β-CD优先与Cl⁻结合,在锌/电解质界面处重新分配阴离子及水分子分布。由此形成富含Cl⁻且界面水可及性降低的结构环境,既能促进瞬时锌–氯相互作用,又能抑制HER及表面钝化现象,同时避免形成强键合的锌–配体复合物。得益于界面调控机制,Zn-Zn对称电池在60mA cm-2下可实现99.8%的高CE;而碱性锌–铁液流电池在80mA cm-2下可稳定运行超过360小时(完成720次循环),峰值功率密度达0.62Wcm-2。
感谢湘潭大学黄志峰老师校稿!
本文所用
螺栓型液流单电池测试夹具(LSB-1)
由武汉之升新能源有限公司提供

《2025年我司用户发表的液流电池论文合集》
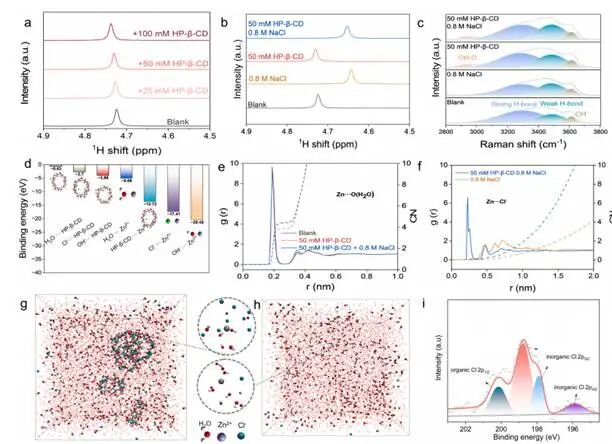
图1对不含HP-β-CD(浓度25至100mM)、氯化钠(0.8 M)及其混合物的0.4M [Zn(OH)₄]²⁻空白电解液进行分子间相互作用研究
¹H NMR谱显示将HP-β-CD加入碱性锌酸盐电解质中仅引起水分子质子共振信号向低场方向轻微移动,表明HP-β-CD与电解质中的氢键网络相互作用较弱,且不会引发整体脱水现象。而且,当同时加入HP-β-CD(50mM)和Cl⁻(0.8M)时,水分子质子的化学位移相对于单独使用Cl⁻的电解质呈现中间值,表明HP-β-CD通过超分子相互作用部分抵消了Cl⁻的结构破坏效应,而非简单叠加两者的贡献。这种偏离简单相加行为的现象提示Cl⁻并非自由分布于电解质中,而是通过HP-β-CD介导发生了空间重分布。此外,这种结合具有动态性和非化学计量特性表明HP-β-CD并未形成刚性的主客体复合物,而是作为Cl⁻的空间组织剂发挥作用。
DFT计算显示Zn²⁺与OH⁻的相互作用最强(−20.45eV),其次为Cl⁻(−17.41eV),与HP-β-CD的相互作用相对较弱(−13.72eV),表明在碱性电解质平衡条件下,Zn²⁺主要以[Zn(OH)₄]²⁻形式存在,而非形成强配位型Zn–HP-β-CD复合物。对于HP-β-CD而言,其与Cl⁻存在中等强度的相互作用(−2.70eV)。能量层级关系排除了基于配位的稳定机制,表明HP-β-CD主要与Cl⁻而非Zn²⁺发生相互作用,从而避免形成动力学反应缓慢的锌–配体复合物。
分子动力学模拟表明在无添加剂条件下,Zn²⁺主要由OH⁻和水配位。单独引入HP-β-CD对Zn–O配位环境的影响可忽略不计,表现为Zn–O_HP-β-CD的配位数极低,证实HP-β-CD不会直接与OH⁻竞争Zn²⁺的配位。相比之下,当同时引入HP-β-CD和Cl⁻时(HP-β-CD-Cl⁻体系),可观察到阴离子在Zn²⁺周围的显著重新分布。而且,在更短距离处(0.23nm)出现了明显的Zn–Cl RDF峰,而该峰在仅含Cl⁻的电解质中(0.48nm)并不存在。Zn–Cl的平均配位数始终较低(<0.2),表明这种相互作用具有瞬时且非化学计量的特性。该峰的存在直接证明了在界面溶剂化环境中,由超分子介导的Cl⁻局部富集所导致的瞬态Zn–Cl相互作用形成概率增加。空间分布分析进一步揭示了Cl⁻在Zn²⁺周围呈现显著富集现象,而这种富集现象在仅含HP-β-CD的电解质中不存在,在仅含Cl⁻的电解质中则明显减弱。此现象源于碱性电解质中由竞争吸附驱动的阴离子重新分布机制。在未添加HP-β-CD时,Cl⁻在电解质主体中随机分布,无法有效参与局部Zn²⁺溶剂化环境;加入HP-β-CD后,通过动态氢键介导的相互作用使Cl⁻向锌界面重新分布,导致局部Cl⁻富集并伴随部分水分子耗尽。上述结果表明存在一种非配位阴离子重分布机制:HP-β-CD更倾向于作为空间调节剂而非配位配体发挥作用。
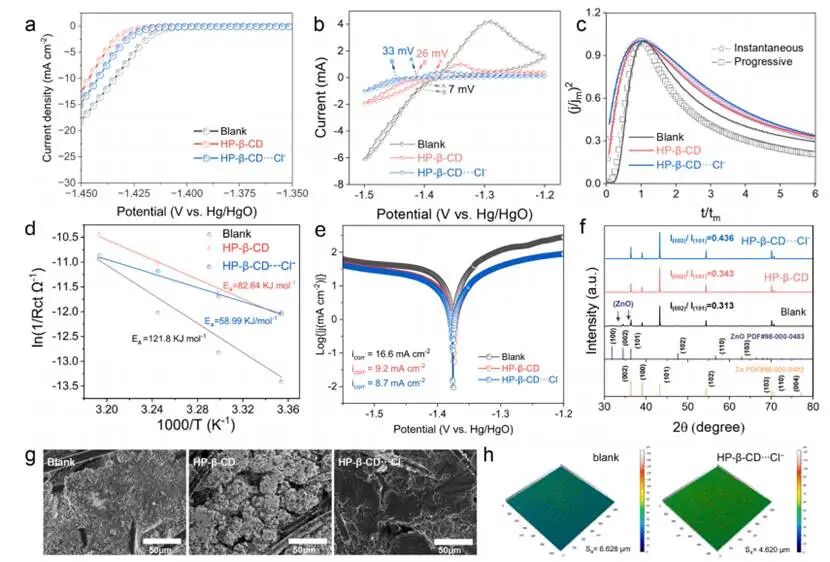
图2.针对不同电解质的锌成核/沉积分析
LSV显示在HP-β-CD-Cl⁻体系中,HER电位相较于空白电解液出现负向偏移,表明该体系的界面水活度降低,与氯离子重新分布导致锌电极表面形成缺水的内亥姆霍兹面相一致。CV数据显示锌核化过电位(η)从空白条件下的7mV上升至仅使用HP-β-CD时的26mV,并在HP-β-CD-Cl⁻存在时进一步升至33 mV,反映出核化过程变得更加受控且均一。
CA测量进一步揭示了从空白电解质中观察到的渐进成核到恒电位(-1.43至-1.40 V)下HP-β-CD⋅⋅·Cl−系统中的瞬时成核的明显转变,表明界面能量的均匀化。温度依赖性EIS分析显示HP-β-CD-Cl⁻体系的活化能(Ea)显著降低至59.0kJ mol⁻¹,较空白电解液(121.8kJ mol⁻¹)下降51%,且明显低于纯HP-β-CD电解液(82.6kJ mol⁻¹)。较低的活化能有助于加速界面电荷转移,同时保持有序的成核过程,从而实现快速、均匀的Zn²⁺沉积并最大限度减少极化效应。塔菲尔分析显示HP-β-CD-Cl⁻体系的腐蚀电流密度(8.7mA cm⁻²)显著低于空白电解液(16.6mA cm⁻²)和纯HP-β-CD电解液(9.2mA cm⁻²),表明重构的界面层通过降低水接触性并稳定电极表面,有效抑制了副反应的发生。
非原位XRD图谱显示空白电解液中形成了氧化锌,表明发生了阳极钝化反应。相比之下,在含HP-β-CD的电解液中这种不利的钝化现象被完全抑制。在控制面容量沉积条件(6.67mAh cm⁻²)下获得的SEM显示:空白电解液中锌沉积物呈不规则且苔藓状,而HP-β-CD-Cl⁻体系则形成光滑致密的锌覆盖层。此外,激光共聚焦扫描显微镜用于表征三种电解质中锌沉积物的表面形态。与空白电解液(6.628 μm)及单独使用HP-β-CD的电解液(5.556μm)相比,HP-β-CD-Cl⁻电解液可实现显著降低的表面粗糙度(4.620μm)。
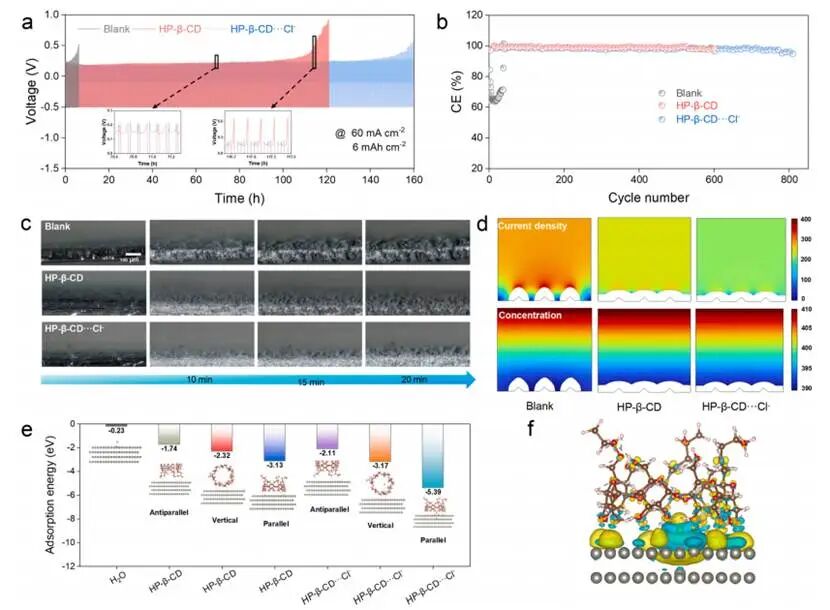
图3.循环性能及机制
使用空白电解液的电池在8小时内即发生失效,而HP-β-CD-Cl⁻体系则能在低极化条件下保持稳定的锌沉积/析出过程约150小时,并展现出高达99.8%的稳定CE。通过原位光学显微镜显示空白电解液中明显的突起结构在5分钟内迅速演变为枝晶结构;虽然仅使用HP-β-CD体系可部分抑制枝晶形成,但HP-β-CD-Cl⁻体系则表现出显著更强的抑制能力,在整个沉积过程中均能保持电极表面光滑均匀。COMSOL软件进行的有限元模拟分析显示在空白电解质中,电场高度集中在突起尖端处,从而促进尖端定向生长。相比之下,HP-β-CD-Cl⁻体系中的电场与Zn²⁺浓度均呈均匀分布,消除了树枝状晶核形成与生长的驱动力。
从头算计算表明水分子的吸附能(Eads)相对较低(−0.23eV),而HP-β-CD的吸附能力显著更强。并且,在HP-β-CD-Cl⁻体系中吸附能进一步提升至−5.39eV。电荷密度差异分析表明在Zn/Cl⁻-HP-β-CD界面处存在局部电荷重新分布,说明Cl⁻优先占据锌表面电子亲和力较高的区域,并通过与HP-β-CD的氢键作用保持稳定。
此外,MD与DFT的综合结果表明存在一种多尺度调控机制:在电解质本体中,HP-β-CD作为非配位阴离子再分配剂,可使Zn²⁺附近Cl⁻浓度升高并降低局部水活度;在电极界面处,HP-β-CD与Cl⁻的协同吸附会置换水分子,形成富含Cl⁻且含水量较低的界面;这种局部结构重构能促进瞬时成核、实现优先沿(002)晶面生长以及获得均匀的电场分布。由于HP-β-CD不会形成强结合的锌–配体复合物,锌酸盐脱羟基反应及电荷转移的能量势垒始终较低。因此,该体系既能增强界面稳定性,又能保持快速反应动力学特性,从而克服了全电池运行中长期存在的稳定性和动力学之间的权衡问题。
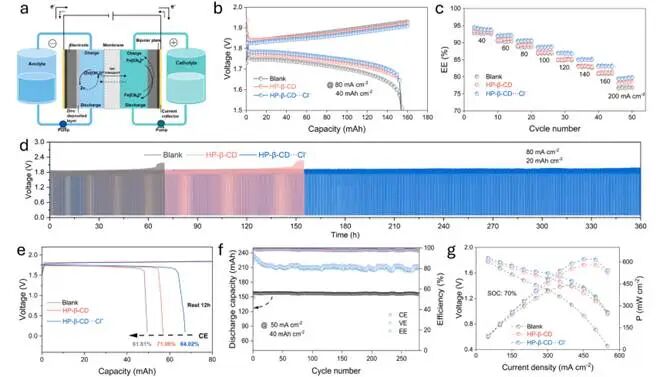
图4使用不同阳极液时AZFBs的电化学性能
在80mA cm⁻²下,HP-β-CD-Cl⁻体系展现出最低的极化电压,表明具有更快的电荷转移动力学。相应地,该电池在40mA cm⁻²下可实现94.2%的高EE,在120mA cm⁻²时仍保持88.7%的EE,优于使用空白电解液或仅含HP-β-CD电解液的电池。而且,在200mA cm⁻²下仍能保持80.2%的EE,远高于现有基于配位化合物的体系。结果表明HP-β-CD-Cl⁻体系有效解决了通常限制配位化合物电解液性能的反应动力学与界面稳定性之间的权衡问题。
长期循环测试显示使用空白电解液的电池性能迅速下降,76小时内即发生失效;而仅采用HP-β-CD电解液时,循环寿命可延长至约140小时。相比之下,HP-β-CD-Cl⁻体系在360小时(720次循环)内保持稳定运行,CE和EE分别维持在95.5%和85.0%。结果证实实现稳定的界面调控关键在于优化阴离子重新分布,而非单纯提高Cl⁻浓度。