
第一作者:杜俊彦
通讯作者:王丽君
通讯单位:北京科技大学
成果简介
钒液流电池(VRFB)被视为大规模储能的核心技术,其中电解液的浓度与稳定性直接影响系统能量密度与运行成本。然而,传统电解液体系普遍存在浓度低、稳定性差以及制备过程复杂等问题。在此,北京科技大学王丽君团队以 VOCl3作为钒源与内生氯供体,并与硫酸组合构建VOCl3-H2SO4电解液体系(2.7 M V,8.1 M Cl–),3.0 M SO42-),该策略可使 VOCl3同步释放钒离子与氯离子,提高钒离子的溶解速率并优化溶剂化结构的稳定性,同时无需外加盐酸。Cl–与 SO42-的协同配位有效抑制沉淀生成。实验结果表明,该电解液在 −5 至 50 °C 的宽温度范围内保持稳定且无沉淀。基于该电解液装配的 VRFB 在 50 °C、80 mA cm-2条件下实现了超过 80% 的能量效率,库仑效率稳定在 96% 以上。
相关成果以“Chloride self-supply electrolyte: Mitigating concentration-stability conflict in vanadium batteries”为题发表在Journal of Energy Storage期刊上。
感谢北科大王丽君团队(第一作者:杜俊彦)供稿!
本文所用
一体化液流单电池测试系统(YTH-1/LSB-1)
由武汉之升新能源有限公司提供


研究背景
VRFB 以溶液形式存储能量,电解液的体积与浓度直接决定电池的储能容量与能量密度。然而,现有电解液中的钒浓度相对较低且热稳定性不足,导致电池性能与成本受限。提升钒电解液性能主要可从钒原料与支持电解质两方面入手。当前,钒电解液主要由 V2O5与VOSO4制备,但V2O5的溶解速率较慢,需在 80~90 ℃条件下加热方能实现高效溶解,这降低了制备效率并增加成本。此外,传统以硫酸为支持电解质的体系也限制了 VRFB 的工作范围。例如,在硫酸体系中,V5+在高于 45 ℃时易以V2O5形式析出,而V3+与V4+在较低温度(<10 ℃)下也易发生沉淀。因此,硫酸体系中钒离子浓度通常受限于2.0 M以下,电池可稳定运行的温度范围也仅限于10~35 ℃。
为解决上述问题,Li等人提出了具有更高钒离子溶解度与更宽工作温度范围的混酸支持电解质。目前已报道的混酸体系包括H2SO4-HCl、H2SO4-H3PO4与H2SO4-CH3SO3H等,其中 H2SO4-HCl混酸体系最为成熟。该体系能够溶解更高浓度的钒离子(>2.0 M),并提供更宽的工作温度范围。在传统硫酸支持电解质中,V5+主要以[VO2(H2O)3]+形式存在,易在升温下转化为VO(OH)3,最终形成 V2O5·3H2O沉淀;而在H2SO4-HCl体系中,钒离子与氯离子络合形成稳定的 VO2Cl(H2O)2物种,有效抑制V2O5沉淀的生成,显著提升高浓度V5+电解液的稳定性,使能量密度由25 Wh L-1提升至40 Wh L-1。Bon 等人进一步通过分子模拟发现,在H2SO4-HCl混酸电解质中,V5+以[VO(OH)2+Cl–SO42-2(H2O)]–络合物存在,同样能够抑制沉淀形成。Yang 等人将V2O5溶解于H2SO4-HCl体系中,当电解液组成为2.4 M钒、6.2 M氯离子与2.5 M硫酸根时,所组装的VRFB可在 −20 ~ 50 ℃范围内稳定运行。综上,H2SO4–HCl混酸体系的应用不仅提高了钒电解液的浓度与能量密度,还降低了与电解液温控相关的系统成本。然而,在以H2SO4–HCl为支持电解质的钒电解液体系中,常用钒源V2O5仍存在溶解度低、溶解速率慢的问题,限制了钒电解液浓度的进一步提升与成本的进一步下降。
核心内容
3.1 使用草酸制备电解液的参数优化
3.1.1 还原剂的选择
本研究选取草酸、甲酸和过氧化氢作为还原剂,主要基于其较强的还原能力、环境友好性以及操作便捷性。三种还原剂参与的还原反应的化学方程式见方程(1)至(3),各反应产物的选择依据相关前人研究。通过查阅文献获取反应物的热力学参数,并据此对相应的还原反应进行热力学分析。相关数据见表 1,计算结果如图1(a)所示。结果表明,三种还原剂对五价钒的还原反应的ΔGΘ均为负值,说明这些反应在热力学上是自发的。其中,草酸参与的反应具有最负的ΔGΘ值,表明其自发性更强、热力学驱动力更大。除此之外,反应(1)的ΔGΘ随温度升高而降低,提示将温度由20 °C提升至100 °C有利于该反应。综合三种还原剂的还原性能,最终选择草酸作为还原剂,并通过化学还原法制备电解液。
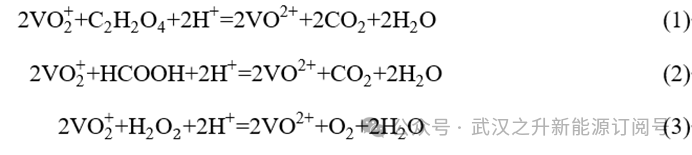
3.1.2 n(C2H2O4):n(VOCl3)比对钒还原率的影响
在反应温度90 ℃、反应时间40分钟条件下,考察了n(C2H2O4):n(VOCl3)比对电解液还原率的影响,结果见图1(b)。随着n(C2H2O4):n(VOCl3)从0.35提高至0.5,电解液的还原率由65.5%升至94.75%。当比例达到0.5:0.57时,还原率出现平台,表明钒的还原基本趋于饱和。进一步分析表明,当n(C2H2O4):n(VOCl3)小于0.5时,加入的草酸量不足,无法将VOCl3中的V5+完全还原为V4+;相反,当该比值大于0.5时,继续增加草酸并不会显著提升还原率,反而会引入草酸杂质,且过量草酸进入电解液会对其电化学性能产生不利影响。因此,n(C2H2O4):n(VOCl3)的最优比例确定为 1:2,这与反应(1)的化学计量比一致。
3.1.3 反应时间对钒还原率的影响
在n(C2H2O4):n(VOCl3)=1:2、反应温度90 ℃的条件下,研究了反应时间对电解液还原过程的影响,结果见图1(c)。电解液的还原率在前20分钟内迅速提升,在40分钟时达到 94.75%。当还原时间延长至100分钟时,还原率变化不大,表明草酸在约40分钟内即可充分还原V5+。为提高制备效率、降低能耗,选取40分钟作为反应时间。图1(c)同时给出了Yang等人报道的反应时间对钒还原率的影响。以草酸还原V2O5时,40分钟的还原率仅为86%,而在100分钟时提升至93.3%。该对比表明,草酸与VOCl3的还原反应速率更快。此外,我们评估了 VOCl3在不同温度下的水中溶解度(见图1(d))。随着温度升高,VOCl3在水中的溶解度先增大后减小,并在40 ℃时达到最大值23.73 g/L。已有研究表明,V2O5在20 ℃时在水中的溶解度仅为1.092 g/L。在相同条件下,VOCl3 在水中的溶解度显著高于V2O5,且由于液–液反应机理,VOCl3的还原速率更快。这显著缩短了还原时间,并减少了高温条件下溶液的挥发,从而提升了整体工艺效率。
3.1.4 反应温度对钒还原率的影响
在n(C2H2O4):n(VOCl3) =1:1的条件下,研究了反应温度对电解液还原过程的影响,结果见图1(e)。随着反应温度升高,还原率逐渐增加。这可归因于较高温度有利于将V5+还原为V4+,与图1(a) 中“升高温度有利于反应”的观察一致。然而,当反应温度提高至97 °C(93.9%)时,与90 °C(93.5%)相比还原率并无显著提升,且出现液体挥发等问题。因此,综合考虑在保证电解液还原率的前提下降低反应温度,以及实验数据与体系稳定性,最终将90 °C选定为最佳反应温度。
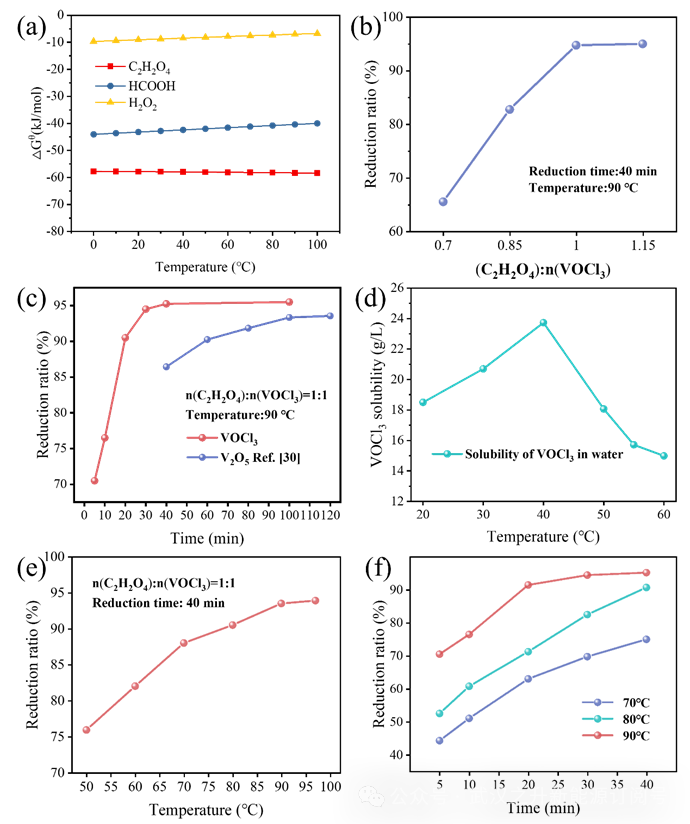
图1. (a)不同还原剂对 V(V) 还原反应的吉布斯自由能变化;(b) n(C2H2O4):n(VOCl3)比对钒还原率的影响;(c)草酸还原VOCl3与V2O5的反应时间对钒还原速率的影响[18];(d) VOCl3在不同温度下的水中溶解度;(e)反应温度对钒还原率的影响;(f)不同温度与不同时刻下电解液的还原率。
3.1.5 还原反应的动力学分析
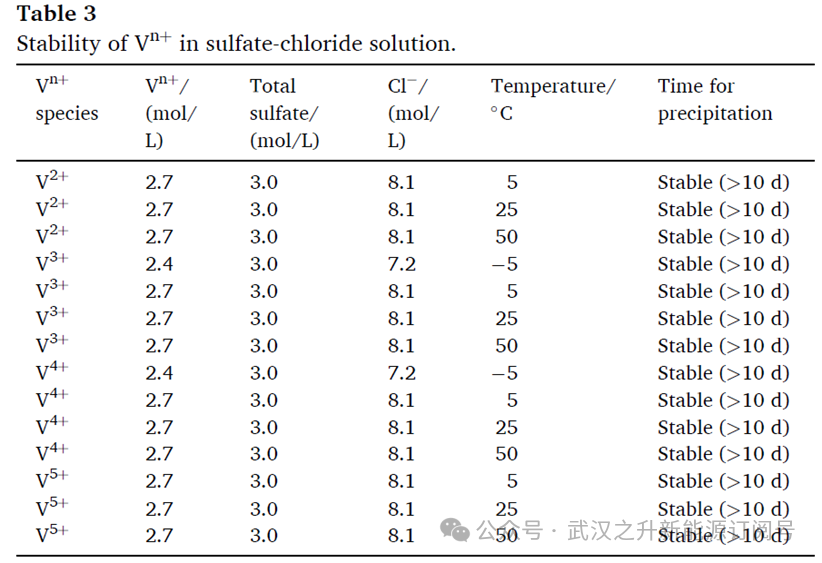
基于还原反应类型,采用拟一级动力学模型对V5+与草酸的还原行为进行模拟(方程(4))。对不同条件下的还原率进行了动力学拟合。图1(f)展示了在70 ℃、80 ℃和90 ℃条件下,不同时间对应的还原率;图2(a)给出了ln(C0/Ct)与反应时间的关系。第二阶段中V(V)的还原受到限制。
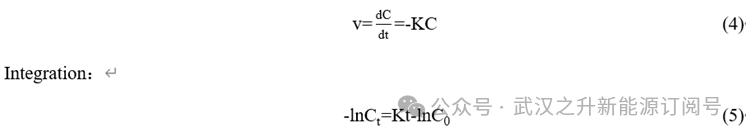
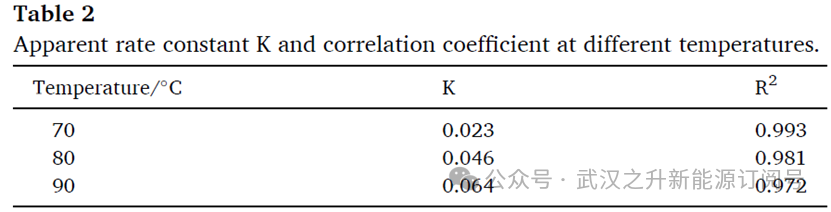
C0与Ct分别定义为实验开始时与给定时间的钒浓度,单位为mol/L。t为时间间隔,单位为 min;K为反应速率常数,单位为s−1。为确定还原反应的常数与活化能,表2汇总了用于拟合方程(5)的表观速率常数K及其相关系数。结果显示,所选模型能够较好地描述还原行为的动力学特征,拟合的R2接近1。将 70 ℃、80 ℃与 90 ℃条件下的数据代入方程(5)拟合(见图2(b)),计算得到钒还原反应的活化能Ea为42.14 kJ/mol。与文献报道的草酸根还原V2O5的活化能73 kJ/mol 相比,本方法中观察到更快的还原速率可从活化能角度得到解释。此外,反应温度、反应时间以及草酸用量在还原过程中发挥协同作用。进一步地,还原时间显著缩短,从而提升制备效率。
3.2 钒电解液的静态稳定性
已有研究报道,在硫酸体系中,钒电解液(< 1.7 M V)的稳定性受限,主要由低温(10 ℃)下VOSO4的低溶解度以及高温(40 ℃)下V2O5的析出所决定。在本研究中,作者在5 °C至50 °C的温度范围内评估了该体系中Vn+的稳定性。测试在无流动、无电流的静态条件下进行,旨在评价电解液在非通电状态下储存与运输过程中的化学稳定性。相关稳定性测试结果见表 3。在含有3.0 M SO42−与8.1 M Cl−的混合溶液中,四种价态的钒在浓度最高至 2.7 M 时均保持稳定。此外,2.4 M 的V3+与V4+电解液在-5 ℃下也保持稳定。鉴于对高温条件下 V5+电解液稳定性的关注,作者进一步考察了2.7 M V5+电解液在50 ℃下的稳定性。经过10天观察,体系中未检测到沉淀,表明V5+电解液具有优异的高温稳定性。更宽的工作温度范围将显著降低或可能消除在大多数地区对电解液温控的需求,从而大幅提升储能效率并降低储能成本。
3.3 钒电解液的溶液化学
3.3.1 由VOCl3制备的钒溶液的稳定性计算
通过计算不同结构的偶极矩与结合能,对各结构及体系的稳定性进行说明与比较。表4给出了不同物质之间的偶极矩,表明偶极矩越大,在极性溶剂中的溶解性越强。可以看出,VOCl3与SO42−之间的配位能力较强,VOCl3-SO42−的偶极矩大于VOCl3-H2O的偶极矩。此外,VOCl3与SO42−的配位会增大 VOCl3 在水溶液中的偶极矩。这一增强可能归因于Cl−与SO42−的络合作用,从而提升了该物质的溶解度。表5展示了各物质之间的结合能。从结合能角度看,VOCl3与水的配位能力明显弱于与SO42−的配位能力。因此,在实际条件下,VOCl3与SO42−的配位是主要的配位方式。VOCl3水解生成V2O5沉淀的反应在该溶液中不易发生。此外,VOCl3与SO42−配位后,其与水的结合能力得到改善,说明Cl−与SO42−的共存可提高其溶解度。
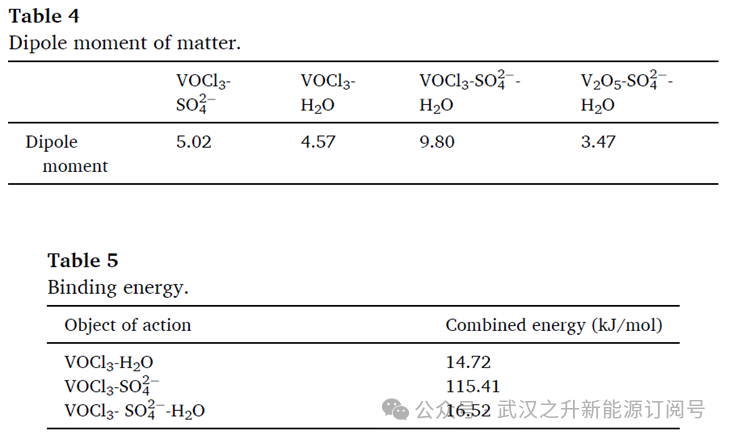
图2(i)和(j)分别展示了在硫酸溶液中形成的VOCl3-H2O和VOCl3-SO42⁻的结构。VOCl3可与SO42⁻形成稳定配合物,这与文献的报道一致。硫酸根的S-O 键与VOCl3中的 V(钒)配位,键长为 1.771 Å,从而使整个钒离子得以稳定。在 V-O-(H2O)结构中,V与SO42⁻仅能形成键长为1.956 Å的配位键。这表明,V 与Cl的配位更有利于分子结构的稳定性。进一步计算显示,V-O-(H2O)与SO42⁻配位后,其S-O键会与配位水分子形成强分子内氢键,键长为1.309 Å;该氢键的形成会显著破坏原有的V-H2O配位结构,导致配合物稳定性下降。然而,V 与氯离子的配位可从根本上抑制这一现象。综上,在由VOCl3提供Cl⁻的硫酸根–氯离子体系中,氯离子的引入更有利于结构稳定。
对体系的 LUMO-HOMO 轨道进行了分析。从图 2(e)和(f)可知,电子从钒的轨道向硫酸根的轨道转移;图 2(g)和(h)则显示,电子从钒的轨道向氯离子的轨道转移。VOCl3中氯离子的存在改变了电子轨道的转移路径,使电子转移距离更短。研究表明,更窄的 HOMO-LUMO 能隙对应更强的得电子能力。因此可推断,VOCl3中的氯离子增强了物质的化学反应活性,使其更有利于V5+的还原。
图2. (a) ln (C₀/Cₜ) 与反应时间的线性拟合图;(b) 还原反应常数与温度倒数的动力学拟合图;优化几何结构:(c) VOCl3-H2O;(d) VOCl3-SO42⁻;(e) V2O5-H2O-SO42⁻;(f) VOCl3-H2O-SO42⁻;LUMO-HOMO轨道图:(g)、(h) 为V2O5-H2O-SO42⁻的 LUMO 与 HOMO 轨道图;(i)、(j) 为VOCl3-H2O-SO42⁻的 LUMO 与 HOMO 轨道图。
3.3.2 钒电解液的拉曼光谱与核磁共振(NMR)光谱分析
鉴于V5+在高温下的不稳定性,本研究对不同温度下V5+的溶液结构进行了表征。首先,采用拉曼光谱分析溶液结构:图3(a)展示了 298 K(25 ℃)和 323 K(50 ℃)下,钒电解液在200–1400 cm⁻1波数范围内的拉曼光谱。不同物种的各类振动模式对应的波数及其归属总结于表6中。根据已报道的钒氧化物拉曼光谱特征,760–790 cm⁻1范围内的谱峰与V–O–V伸缩振动相关。该谱峰的出现源于溶液中较高的钒浓度(>1.75 mol/L),表明高浓度下溶液中存在V5+二聚体。拉曼光谱结果显示,溶液中同时存在单核钒物种和双核钒物种。随后,通过基于广义梯度近似(Generalized Gradient Approximation,GGA)的密度泛函理论(DFT)计算,得到了(VO2Cl·2H2O)2-(VO2OH)2和[V2O3Cl2·6H2O]2+两种物种的结构(图 3(b))。因此,本研究提出:该电解液中V5+稳定性提升的原因是,VOCl3引入的Cl⁻促使溶液中形成了可溶性中性物种(VO2Cl·2H2O)2-(VO2OH)2和(VO2Cl·2H2O)2-(VO2OH)2,从而抑制了沉淀的生成。
为验证上述假设的可能性,本研究对V5+电解液进行了51V NMR和35Cl NMR测试。图 4(b)–(d)展示了不同温度下电解液的51V NMR和35Cl NMR谱图,谱峰变化趋势具有高可逆性,表明该体系中V5+阳离子具有良好的热稳定性。已有报道指出,在硫酸盐溶液中,[V2O3·8H2O]4+结构的51V谱峰出现在-500至-600 ppm范围内,且中心位于-563 ppm处。与之相比,本研究中电解液的51V谱图显示出一个宽峰,范围为-50至-1000 ppm,中心位于-540 ppm处,该宽峰可能代表双核物种[V2O3·8H2O]4+与钒–氯配合物的混合物。该电解液的51V谱图在化学位移和峰宽上,与硫酸盐溶液中的51V谱图存在显著差异,表明两种溶液具有截然不同的化学环境。Li 等人曾报道,在高浓度混合酸溶液中,[V2O3·8H2O]4+与 [V2O3Cl2·6H2O]2+可能共存;但本研究推测,NMR谱图中还可能存在另一种钒–氯化合物。
为进一步确定本研究中电解液的结构,作者在不同温度下进行了35Cl NMR 测试。不同温度下电解液的35Cl 谱图(见图3(c)、(d))显示,溶液中存在三种不同的氯成键环境。在 298 K 时,电解液的主峰出现在约 90 ppm 处,这与 H₂SO₄-HCl 体系的35Cl谱峰特征一致;而在约185 ppm处出现的新峰,表明溶液中形成了一种新的含氯成键化合物。在323 K时,谱图中约310 ppm处出现一个与文献报道特征相符的谱峰,该峰可归属于[V2O3Cl2·6H2O]2+;同时,185 ppm 处的谱峰仍保持存在。因此,本研究推测溶液中可能共存两种含氯成键化合物:185 ppm 处对应的(VO2Cl·2H2O)2-(VO2OH)2,以及 310 ppm 处对应的[V2O3Cl2·6H2O]2+。综上,51V和35Cl NMR 测试中新增谱峰的出现,明确支持了本研究的假设–钒电解液中形成了[V2O3Cl2·6H2O]2+与(VO2Cl·2H2O)2-(VO2OH)2两种含氯成键化合物(见图3(e)、(f))。根据 NMR 结果可知:298 K 时,体系中主要存在(VO2Cl·2H2O)2-(VO2OH)2;而在较高温度(323 K)下,含氯成键化合物[V2O3Cl2·6H2O]2+也成为溶液中的重要组分。因此,该体系中51V热稳定性提升的原因,可能是这些新型含氯成键化合物的形成抑制了51V以V2O5·3H2O形式析出沉淀。

图3. (a) 2.4 mol/L(M)V (V) 电解液的拉曼光谱图;在 11.7 T(特斯拉,磁场强度单位)磁场下测得的 2.4 mol/L V (V) 电解液的变温 NMR 分析:(b) 51V NMR 谱图;(c)、(d) 35Cl NMR 谱图;彩色线条代表采用洛伦兹型峰进行的分峰拟合分析。
3.4 钒电解液的电化学性能
本研究在10 mVs-1的扫描速率下,对含3.0 mol/L硫酸根的正极电解液,在不同钒浓度(1.8–2.7 M)和氯离子浓度(5.6–8.1 M)下的 CV 曲线进行了测试(见图4)。如图 5(a)、(b)所示:当钒浓度从 1.8 M 增至 2.4 M、氯离子浓度从 5.4 M 增至 7.8 M 时,正极电解液的反应动力学性能逐步提升;但当钒浓度达到 2.7 M、氯离子浓度达到 8.1 M 时,阳极峰值电流(Ipa)与阴极峰值电流(Ipc)均出现轻微下降,这一现象可归因于反应物浓度与电解液粘度的耦合效应。具体而言:虽然提高钒浓度能提供更多活性反应物,但电解液粘度会随钒和氯离子浓度的增加而升高;粘度增大不仅阻碍离子迁移,还会加剧欧姆极化与浓差极化(两种导致电化学反应效率下降的极化现象)。
为进一步探究钒浓度对电解液电化学行为的影响,本研究采用电化学阻抗谱(EIS)进行分析(见图4(c)、(d))。奈奎斯特图显示,电解液中V4+/V5+的氧化还原反应同时受高频区电荷转移过程与低频区扩散过程控制。研究采用图4(c)所示的简化等效电路模型对奈奎斯特图进行拟合,拟合参数也列于该图中。结果表明:溶液电阻与电荷转移电阻均随钒和氯离子浓度的升高而增大。这是因为随着离子浓度增加,正、负离子间的相互作用增强,导致离子迁移速率降低。此外,奈奎斯特图低频区的电阻值反映离子在电解液中的扩散过程:扩散阻抗(W)随钒和氯离子浓度的升高呈现“先增后减”的趋势,说明钒与氯离子浓度的变化对低频区离子传输过程存在显著影响。综上,CV 与 EIS 测试结果均表明,在VRFB中使用高浓度钒电解液具有优势。如图4(e)、(f)所示:随着硫酸浓度升高,Ipa与Ipc均逐步增大。这是因为H+迁移速率较快,较高的硫酸浓度可提升电解液电导率,进而促进电化学反应;但同时,硫酸浓度升高也会导致电解液粘度增大,对离子迁移产生不利影响。上述两种因素的相互作用,最终形成了Ipa与Ipc的变化趋势。图4(g)为不同硫酸浓度下电解液的 EIS 数据:当硫酸浓度从 2.4 M 增至 3.0 M 时,溶液电阻逐步降低,这可能是由于H+迁移速率加快,使电解液电导率提升;而当硫酸浓度从 3.0 M 增至 3.3 M 时,扩散阻抗显著增大–这是因为电解液黏度随硫酸浓度升高而增加,阻碍了钒离子的迁移。
综上,虽然较高的钒浓度对电池性能有利,但硫酸浓度过高导致的黏度增大会降低 VRFB 的运行效率。因此,理想的操作条件应是在适宜浓度的硫酸溶液中维持较高的钒浓度。基于此,本研究最终选定的电解液浓度为:2.7 M 钒、8.1 M 氯离子、3.0 M 硫酸。
图4.(a)、(b)不同硫酸根浓度下(钒浓度 2.4 mol/L、氯离子浓度 7.2 mol/L),正极电解液在 10 mVs-1扫描速率下的循环伏安(CV)曲线,及对应的Ipa、Ipc与ΔEp;(c)V5+/V4+氧化还原对的奈奎斯特图(Nyquist 图)及等效电路;(d)、(e)不同硫酸根浓度下(钒浓度 2.4 mol/L、氯离子浓度 7.2 mol/L),正极电解液在 10 mVs⁻1扫描速率下的循环伏安(CV)曲线,及对应的Ipa、Ipc与ΔEp;(f)V5+/V4+氧化还原对的奈奎斯特图(Nyquist 图)及等效电路。
3.5 含该钒电解液的VRFB性能
本研究选取钒浓度 2.7 mol/L、硫酸根浓度 3.0 M、氯离子浓度 8.1 M 的电解液进行单电池性能测试,在 – 5~50 ℃温度范围及 40~120 mAcm-2电流密度下开展充放电实验。如图5(a)、(b)所示,25 ℃时:CE随电流密度升高而增大,这是因为更高的电流密度会缩短充放电时间,从而减少钒离子透过隔膜造成的容量损失;与之相反,VE随电流密度升高而降低,原因是电流密度越大,电解液中的欧姆极化越显著;EE同样随电流密度升高而下降,在电流密度为 40 mAcm-2时,EE 达到 81.6%。图5(b)显示,随着电流密度增大,充电电压平台升高、放电电压平台降低–这一趋势与电化学测试中观察到的欧姆极化、浓差极化及电化学极化变化规律一致。此外,如图5(b)所示,极化程度随电流密度升高持续加剧,这与 EE 的变化趋势相符。
图5(c)展示了电流密度为 80 mAcm-2时,单电池在 – 5~50 ℃温度范围内的性能。由于高温下钒离子透过隔膜的渗透率更高,CE 随温度升高略有下降;而 VE 随温度升高逐步增大,这是因为高温下电解液电导率更高、黏度更低,可减少浓差极化与欧姆极化;EE 也随温度升高而提升,在25 ℃时达到 84.3%,50 ℃时达到 85.2%。Yang 等人曾报道过相似条件下的单电池性能:其采用的电解液含2.4 M 钒、2.5 M 硫酸根及6.2 M 氯离子,结果显示该体系的 CE 为 96.21%,高于V2O5体系的91%;当电流密度在40~80 mAcm-2范围内时,本研究中2.7 M钒体系在50℃下的EE(85.2%)优于2.4 M V2O5体系的EE(77%)。这些结果表明,使用该电解液的VRFB在高温下仍具有优异的能量效率。图5(d)为电流密度 80 mAcm-2时,-5~50 ℃温度范围内的充放电曲线。随着温度升高,平均充电电压降低、平均放电电压升高–通过提高工作温度可减小 VE 极化,进而降低充放电过程中的过电位,这与图5(a)、(c)所示的循环效率结果一致。此外,图5(e)为 25 ℃、80 mAcm-2电流密度下的550次循环测试结果。550次循环后,电池的能量效率仅下降0.78%,且整个测试过程中电池运行稳定,未出现任何沉淀。

图5. (a)25 ℃下,不同电流密度下VRFB的CE、VE与EE;(b)25℃下,不同电流密度下 VRFB 的充放电曲线;(c)80 mAcm-2电流密度下,不同温度下 VRFB 的库仑效率CE、VE与EE;(d)80 mAcm-2电流密度下,不同温度下 VRFB 的充放电曲线;(e)25 ℃、80 mAcm-2电流密度下,使用 2.7 mol/L(M)钒电解液的 VRFB 在 550 次循环过程中的CE、VE与EE。
结论展望
本研究以VOCl3为原料,将其直接溶解于硫酸溶液中,并利用VOCl3提供的氯离子,制备出基于硫酸根-氯离子复合体系的钒电解液。主要研究结论如下:
- 还原反应最优条件:还原反应的最佳条件为—— 反应温度 90 ℃、反应时间 40 分钟、草酸与VOCl3的摩尔比 1:1。在此条件下,钒的还原率可达 95%。
- 体系中SO42⁻与Cl⁻的共存可抑制沉淀生成,并提升体系的氧化还原能力。2.7 mol/L的钒电解液在-5 ℃~50 ℃温度范围内保持稳定,这得益于与V5+相关的两种钒-氯化合物的形成,该类化合物可有效阻止沉淀产生。
- 基于电化学性能测试,确定优化后的电解液浓度为–钒2.7 M、硫酸根3.0 M、氯离子8.1 M。
- VRFB 性能测试表明,单电池在-5 ℃~50 ℃温度范围内可稳定运行。在25 ℃、电流密度80 mAcm-2的条件下,2.7 M 钒电解液对应的EE达到 85.2%。
文献信息
Junyan Du, Huitong Lin, Longyan Zhang, Shiyuan Liu, Lijun Wang, Chloride self-supply electrolyte: Mitigating concentration-stability conflict in vanadium batteries. Journal of Energy Storage, 2025, 134: 118098.
https://doi.org/10.1016/j.est.2025.118098
延伸阅读(王丽君团队近期论文合集):
【液流电池论文赏析】祝贺我司客户北科大王丽君团队发表CEJ: 新型电解液设计增强低温下3.0MV3+的稳定性用于高效钒液流电池
【液流电池论文赏析】祝贺我司客户北科大王丽君团队发表JES:一种绿色、经济高效的高纯度VOCl3制备方法及其在钒液流电池中的应用
【液流电池论文赏析】最新AFM(IF=18.5)综述!钒氧化还原液流电池的先进材料:主要障碍和优化策略