
第一作者:林柳
通讯作者:李喆珺,陈胜利
通讯单位:武汉大学
DOI:10.1016/j.joule.2026.102434
感谢武汉大学李喆珺团队(第一作者:林柳)校稿!
成果简介
基于多硫化物的水系氧化还原液流电池(PSARFBs)由于多硫化物还原动力学缓慢,存在功率密度方面的固有局限。本研究揭示此现象源于多硫化物在双电层(EDL)内向带负电荷电极迁移的过程受到抑制。并且通过引入痕量1-乙基-1-甲基吡咯烷鎓(EMP⁺)作为阳离子电催化剂,显著促进了多硫化物迁移,将反应速率常数提升了两个数量级。EMP⁺通过可逆的界面吸附作用机制屏蔽局部负电荷而不发生化学转化,实现持久的动力学调控。采用EMP⁺的多硫化物负极电解液展现出良好的电化学稳定性(1,300次循环/64.5天),且体积容量可高达100 Ah L⁻¹。经6 mM EMP⁺催化的PSARFBs在150 mA cm⁻²下表现出优越的倍率性能:容量利用率达99.3%。阳离子催化PSARFBs可稳定循环超过2000次,容量衰减速率仅为0.000033%/循环(0.0014%/天)。该研究为提升PSARFBs的功率性能提供了理论基础。
背景分析
为提升PSARFBs的功率性能,研究者们投入了大量精力通过引入异相和均相催化剂来加速多硫化物的氧化还原反应动力学。例如,金属硫化物(CoS、CuS、WS₂、NiCo₂S₄等)以及单金属原子(Co、Ni等)等固态催化剂已被广泛用作潜在催化剂候选物,以降低缓慢的多硫化物氧化还原反应的能量势垒。然而,由于这些催化剂的比表面积有限、电子导电性较差且催化活性较低,研究人员开发了多种微观结构优化策略,包括构建多孔框架、添加导电碳材料,以及调控局部电子结构等。尽管这些策略均旨在改善电荷转移动力学,但尚不清楚这是否是涉及多个步骤的复杂多硫化物氧化还原反应中的限速步骤。因此,为克服非均相/均相催化剂的局限性并进一步释放PSARFBs的功率潜力,需要基于对水介质中复杂多硫化物氧化还原反应机制的深入认知,开发一种新型调控策略。
图文解析
1.揭示多硫化物缓慢还原反应的限制因素
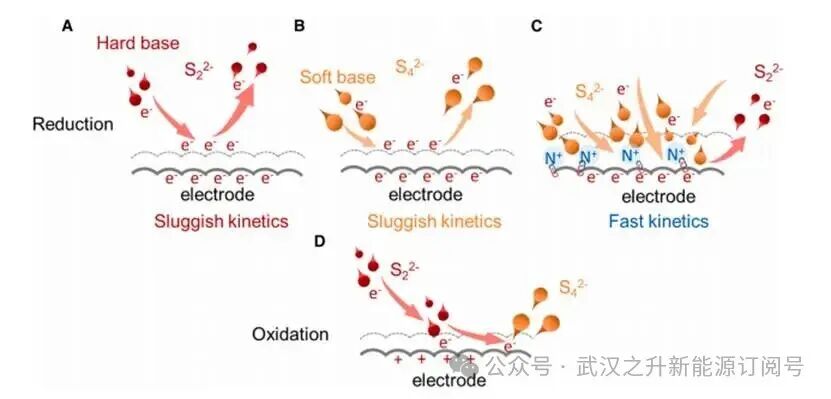
图1.多硫化物还原与氧化过程的不对称行为
高负电荷反应物的还原速率对双电层效应极为敏感,双电层效应会将反应物排斥于电极界面之外,进而延缓还原动力学。由于带负电的电极界面产生的强烈静电排斥作用,路易斯硬碱S22-的还原反应被抑制。而长链反应物S42-(即路易斯软碱)的电荷密度更低,因此这种静电排斥作用较弱。与复杂的还原过程相比,Sx2-的氧化过程面临的迁移阻碍较少,因为电极表面带正电荷。电极带电属性的差异导致多硫化物在水介质中表现出不对称的氧化还原行为。本文开发了一种基于界面工程的阳离子电催化剂,其可在内亥姆霍兹平面(IHP)内自组装,屏蔽电极负电荷,从而降低多硫化物还原过程的迁移势垒。
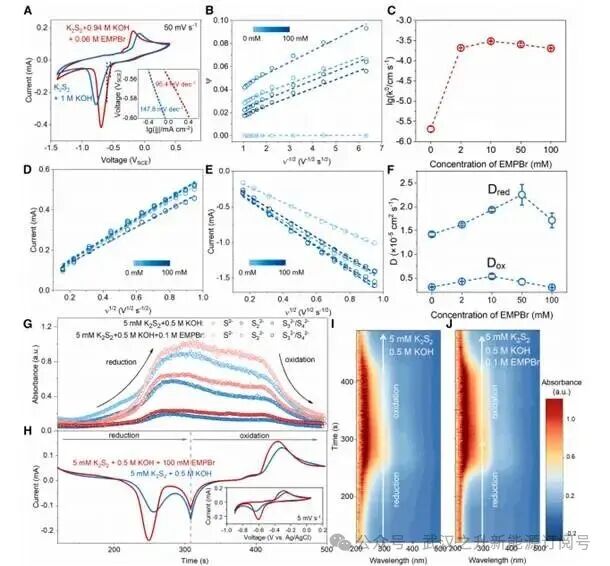
图2.阳离子电催化剂对多硫化物氧化还原反应的促进作用
EMP+可以显著增强多硫化物的反应动力学,尤其对于还原过程。循环伏安法(CV)显示阴极峰与阳极峰的间距在加入60 mM EMP+后从692.4 mV显著减小至508.8 mV,且阴、阳极峰值电流均呈现不同程度的提升。此外,Tafel还原曲线的斜率从147.8 mV dec⁻¹降至95.4 mV dec⁻¹,这表明驱动相同程度多硫化物还原所需的过电位降低。加入EMP+后,形式电位仅发生可忽略的偏移(5.9 mV),表明多硫化物的热力学稳定性不受EMP+存在的影响。
通过分析与拟合不同扫速下的CV数据,可计算不同EMP+添加用量对多硫化物扩散系数和反应速率常数的影响。首先,随着EMP+的浓度从0 mM增加到50 mM,Dred从1.42×10⁻5 cm² s⁻¹逐渐增大到2.26×10⁻5cm² s⁻¹,后又降低为1.72×10⁻5cm² s⁻¹。在低浓度(2–50 mM)条件下,EMP⁺浓度的升高会促进其在IHP中的特异性吸附,使ϕ2电位正移,从而降低电极对Sx²⁻的排斥作用。然而,在较高浓度(100 mM)下,由于特异性吸附的EMP+在电极表面的聚集导致电极表面被阻塞,扩散速率反而降低。此外,与还原过程相比,Dox(约为4.0×10⁻⁶cm² s⁻¹)受EMP+浓度差异的影响较小,这可归因于氧化过程中电极本身带正电荷。反应速率常数的计算结果显示当EMP⁺浓度从0 mM增至10 mM时,k0值从2.06×10⁻⁶ cm s⁻¹增加至3.06×10⁻⁴ cm s⁻¹。由于吸附的EMP⁺会阻塞电极表面,当EMP⁺浓度进一步从10 mM增至100 mM时,k0反而降低。
含/不含EMPBr条件下的原位紫外-可见(UV-vis)吸收光谱显示,与空白电解液相比,EMP⁺催化电解液在还原过程中产生了更高浓度的低价多硫化物物种,表明EMP⁺催化具有更高的还原效率。然而,空白电解液与EMP⁺催化电解液的阳极过程中吸收度变化的斜率相当,这说明EMP⁺对多硫化物氧化过程的影响有限。EMP⁺催化剂显著改善了还原和氧化过程中S²⁻与S2²⁻信号强度的对称性,这可归因于多硫化物还原速率的加快。
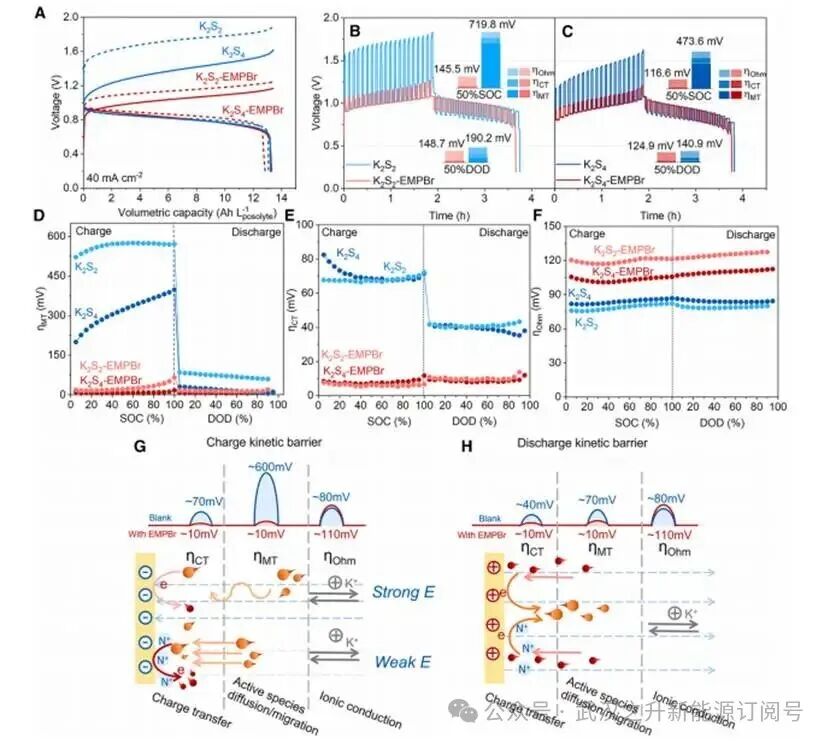
图3.PS-Fe电池的恒电流充放电特性
在40 mA cm⁻²下,多硫化物-亚铁氰化物(PS-Fe)电池充电电压从K₂S₂(1.72 V)和K₂S₄(1.37 V)显著降至K₂S₂-EMPBr(1.14 V)和K₂S₄-EMPBr(1.04 V),相应EE从46.4%和57.9%提升至71.4%和77.1%。GITT结果显示四种电解液具有相似的准平衡电位,这表明多硫化物的热力学特性未受EMP+添加的影响。然而,空白电解液与EMP+催化电解液表现出显著的动力学极化差异,该差异在充电过程中尤为明显。GITT-EIS技术分析显示,物质传输过电位ηMT被明确认定为空白K₂S₂和K₂S₄电解液充电极化的主要贡献因素。例如,K₂S₂电解液在50%SOC下观察到高达573.8 mV的ηMT,占ηtotal的79.7%;而在K₂S₄电解液中该数值降至321.1 mV,占比为67.8%,这归因于带负电电极界面对较软阴离子S₄²⁻产生的静电排斥作用更弱。加入EMP⁺电催化剂后,K₂S₂和K₂S₄的ηMT在50%SOC时分别降至21.0 mV和7.3 mV,降幅分别高达96.3%和97.7%。在负极电解液中,带高负电荷的电极表面在充电过程中会排斥接近的电活性多硫化物阴离子,导致多硫化物的传质效率低下。然而,EMP+的存在可屏蔽负电荷,显著降低传质势垒,从而提高Sx2−向电极附近的迁移速率。此外,由于EMP+特异性吸附产生的ϕ2效应,两者的电荷转移过电位ηCT均降低了约60 mV。
放电过程中,电极表面带正电荷,此时多硫化物发生氧化反应。使用四种负极电解液的PS-Fe电池的放电电压和过电位受多硫化物链长长短或EMPBr存在与否的影响较小。对GITT过电位的分解进一步表明对于空白K2S2和K2S4电解液,放电的ηMT远低于充电过程,这表明放电过程中多硫化物迁移面临的阻碍比充电过程少。
2.通过阳离子电催化剂对EDL的结构优化
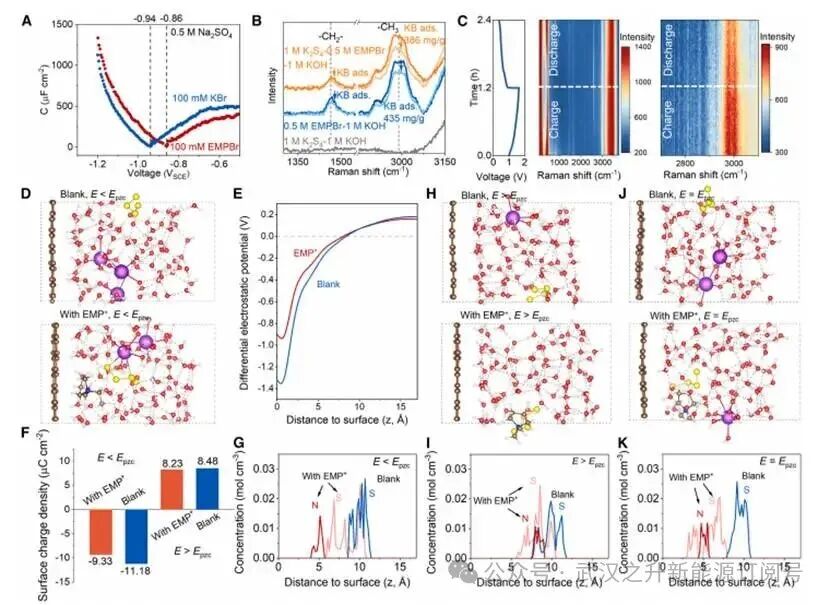
图4.以EMP+作为阳离子电催化剂的EDL结构
交流伏安法显示在EMP+存在下,零电荷电位(PZC)正向移动了80 mV,表明EMP+在IHP中发生了特异性吸附。碳材料KB吸附前后电解液的拉曼光谱证实EMP+在KB表面具有良好的吸附能力,且多硫化物吸附对其影响可忽略不计。原位拉曼光谱对EMP+在一个恒电流充放电循环过程中的动态吸附行为的监测结果显示在充电过程中,2951 cm⁻¹处EMP+的特征性甲基峰的信号强度强于放电过程,表明EMP+在电极上的特异性吸附对施加电势极为敏感,并且在较低电势下、电极带有更多负电荷时吸附作用会增强。
从头算分子动力学模拟显示在负偏压下(即E<Epzc),EMP+可吸附于距电极表面0-5 Å范围内,使表面电荷密度从-11.18 μC cm−2降低到-9.33 μC cm-2,并使EDL处的电势从-1.35 V升高到-0.94 V,促使S42−的分布更接近电极表面(从10.7到6.9 Å)。相比之下,在正偏压条件下(即E>Epzc),EMP+会从电极表面解吸(分布于约8.4 Å处),且几乎不对界面电荷密度(8.23 vs.8.48 μC cm⁻²)和S42−的分布产生显著影响。总体而言,在S4²⁻还原过程中,EMP+优先吸附并聚集于IHP中,以中和排斥多硫化物阴离子的强负电荷,从而相较于无EMP+时显著改善还原动力学。相反,在氧化过程中,EMP+会被带正电的电极表面排斥,因此对氧化速率影响可忽略不计。零偏压(E=Epzc)下对EMP+具体吸附行为的研究进一步表明,尽管缺乏静电驱动力,EMP+仍保持与负偏压条件下相似的吸附位置,说明EMP+在IHP中的吸附行为本质上也受其分子疏水特性的调控。
3.PSARFBs的恒电流性能
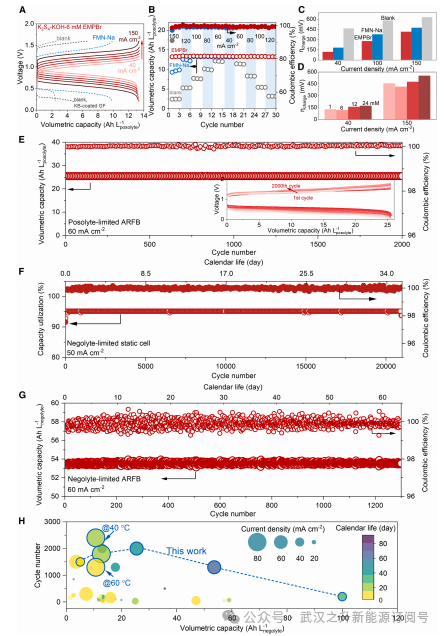
图5.PSARFBs的长期循环
长循环测试中,PS-Fe ARFBs采用电荷增强离子选择性(CRIS)膜。在80 mA cm⁻²下,正极容量限制的催化型PS-Fe ARFB在经过1800次循环(20天)后仍保持99.9%的容量,其容量利用率高达95%,且容量衰减速率极低(为每循环0.000058%)。相比之下,在相同电位范围内,未催化ARFB仅能利用51.65%的容量。此外,通过在负极电解液中使用混合阳离子(K⁺和Na⁺),可将容量提升至25.5 Ah L⁻¹。经过2,000次循环后,该电池容量衰减可忽略不计(衰减速率为0.000033%/循环),这表明该策略在高能量密度ARFB应用中具有广阔前景
核心结论
本研究揭示多硫化物缓慢的氧化还原动力学受限于还原反应过程中带强负电荷的Sx²⁻向带负电荷电极界面迁移受阻。EMP⁺因在电极上的特异性吸附能力而被用作阳离子电催化剂,其可屏蔽电极负电荷,通过ϕ2效应提升多硫化物迁移速率并使反应速率常数提升了两个数量级。这种界面调控通过非消耗性吸附–解吸机制实现,无需化学转化,从根本上避免了催化剂降解,即使在痕量浓度下也能实现持久的动力学调控。所开发的EMP⁺催化多硫化物负极液在实验室条件下表现出优异的耐久性(1300次循环/64.5天)和100 Ah L⁻¹的高体积容量。采用6 mM EMP⁺作为阳离子电催化剂的PS-Fe ARFB实现了高达150 mA cm⁻²的优异倍率输出,容量利用率达99.3%。此外,采用CRIS膜的6 mM EMP⁺催化PS-Fe ARFB在2000次循环中展现出高循环稳定性,容量衰减速率低至0.000033%/循环(0.0014%/天)。