
通讯作者:王珏
通讯单位:中南大学
DOI:10.1016/j.cej.2026.179090

本研究通过基于过渡金属酞菁化合物构建催化中心的电子结构模型,以提升还原反应动力学并提高效率。本研究首次揭示了实现高效S/Fe RFBs所需催化中心电子结构的本质特征,并提出了一种新型描述符来量化电子结构与催化活性之间的关联,成功识别出具有最高催化活性的电子结构构型。在负载具有最佳电子结构的催化剂后,80mA/cm2下的平均EE达到70.70%,比原始电池(45.18%)高25.52%,循环性能在60mA/cm2下从200次循环扩展到1000次循环。
《2025年我司用户发表的液流电池论文合集》
1.理论计算
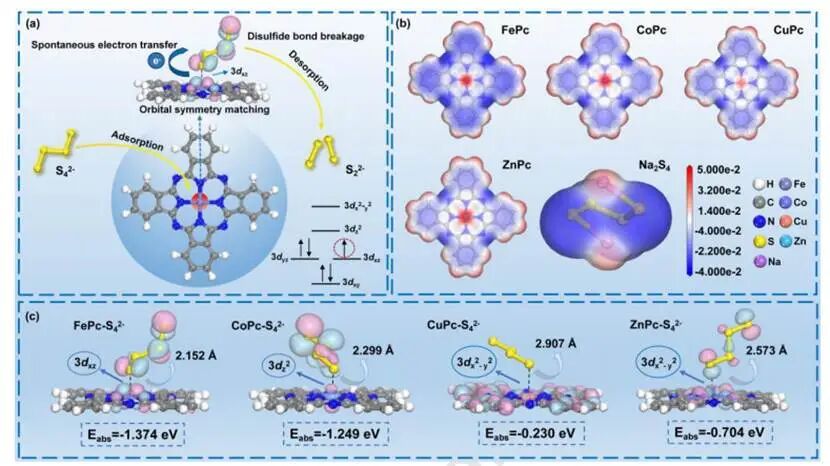
图1(a)基于过渡金属酞菁化合物催化S4²⁻还原的机制;(b)FePc、CoPc、CuPc、ZnPc及Na2S2的静电势分布;(c)FePc-S4²⁻、CoPc-S4²⁻、CuPc-S4²⁻和ZnPc-S4²⁻的吸附模拟及轨道对称性匹配图
与氧化过程的负自由能(−0.91eV)相比,S₄²⁻还原为S₂²⁻所需的能量仅为0.91eV,此非自发过程限制了氧化还原反应的速率。同时,塔菲尔曲线进一步证明由S₄²⁻向S₂²⁺转化引发的缓慢电化学动力学过程主导了极化行为。经确定关键步骤后,S₄²⁻还原为S₂²⁺的过程可划分为四个阶段:S₄²⁻在催化剂表面的吸附、电子从催化剂向S₄²⁻的转移、二硫键的断裂以及S₂²⁺的脱附,均与催化中心的电子结构密切相关。
静电势分布图显示位于M-N₄位点的中心金属离子具有最大的正静电势,而S₄²⁻中末端硫原子的静电势则为最负值,表明由于静电吸引力的作用,酞菁分子内的Fe³⁺、Co²⁺、Cu²⁺和Zn²⁺均是S₄²⁻的吸附位点。此外,在电子结构中,催化中心和目标反应物之间的轨道对称性匹配对吸附有显著影响。催化中心轨道与反应物轨道之间更佳的对称匹配能够实现稳定的反应物吸附,并促进后续电子转移。吸附能(Eabs)计算显示在CuPc-S₄²⁻和ZnPc-S₄²⁻吸附体系中,末端硫原子的2p轨道与Cu²⁺或Zn²⁺的3dx²-y²轨道均不匹配,导致S₄²⁻在Cu²⁺或Zn²⁺表面的吸附不稳定,其吸附能分别为-0.230eV和-0.704eV;而在CoPc-S₄²⁻体系中,Co²⁺的3dz²轨道对称性与硫原子的2p轨道对称性相匹配,表明CoPc吸引S₄²⁻的能力强于CuPc和ZnPc,吸附能为-1.249eV;对于FePc-S₄²⁻体系,Fe³⁺的3dxz轨道与硫原子2p轨道间的π–π配位比Co²⁺的3dz²轨道与硫原子2p轨道间的轴向配位具有更强的轨道重叠,从而实现更稳定的吸附并产生更负的吸附能(Eabs=-1.374eV)。
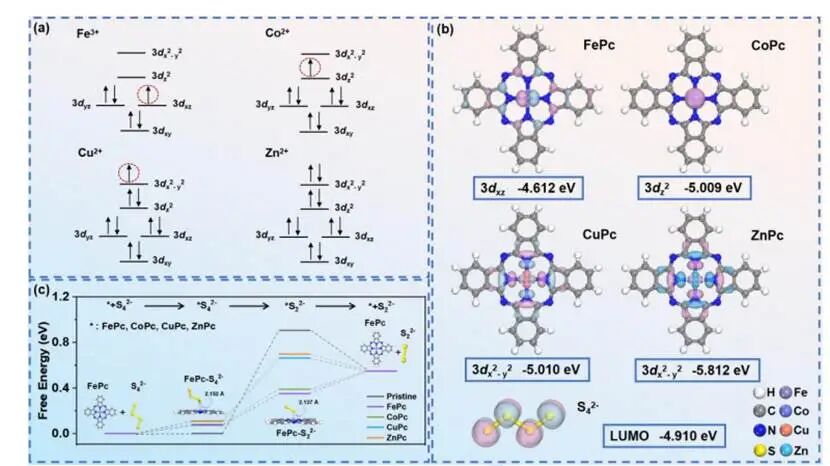
图2.(a)FePc、CoPc、CuPc和ZnPc中位于M-N4位点的中心金属离子在D4h对称性下的电子排布;(b)FePc的3dxz轨道能级、CoPc的3dz²轨道能级、CuPc的3dx²-y²轨道能级、ZnPc的3dx²-y²轨道能级以及S⁴²⁻的LUMO轨道能级;(c)S4²⁻在FePc、CoPc、CuPc和ZnPc上还原反应的自由能变化
由于未配对电子的活性通常高于配对电子,因此从理论上讲,当电子结构中存在未配对d电子时,催化活性会更强。根据FePc、CoPc、CuPc和ZnPc中中心金属离子的d电子排布情况,Zn²⁺的所有d轨道电子均呈自旋配对;相反,Cu²⁺的3dx₂-y₂轨道、Co²⁺的3dz₂轨道以及Fe³⁺的3dxz轨道上均存在未配对电子,比Zn²⁺具有更高的催化活性。在吸附过程稳定且存在未配对电子的情况下,需重点考虑电子转移的自发性:当催化中心含未配对d电子的d轨道能级高于目标反应物S₄²⁻的LUMO轨道能级时,d轨道电子即可自发转移至S₄²⁻。计算结果表明Co²⁺的3dz₂轨道能级为-5.009eV,低于S₄²⁻的LUMO轨道能级,意味着CoPc中的Co²⁺d电子不会自发转移至S₄²⁻;而FePc中Fe³⁺的3dxz轨道能级为-4.612eV,比S₄²⁻的LUMO轨道(2p轨道)能级高出0.298eV,表明存在自发电子转移的可能性。
此外,S4²⁻向S₂²⁻转化过程中二硫键断裂所需的自由能(Ebond)以及随后S₂²⁻解吸附所需的自由能(Edes),并构建了整个催化过程的自由能曲线。计算结果表明在FePc、CoPc、CuPc和ZnPc催化剂体系中,二硫键断裂始终是整个还原反应过程中的限速步骤:FePc催化剂的Ebond值为0.28eV,CoPc为0.32eV,CuPc为0.56eV,ZnPc为0.59eV,均高于上述催化剂上S₂²⁻解吸附所需的自由能;而Edes值则分别为:FePc为0.19 eV,CoPc为0.16 eV,CuPc为-0.12 eV,ZnPc为-0.15 eV。此外,通过比较Ebond可知,FePc在二硫键断裂自由能方面表现出最大的降低幅度,表明具有最佳催化性能。
2.材料特性
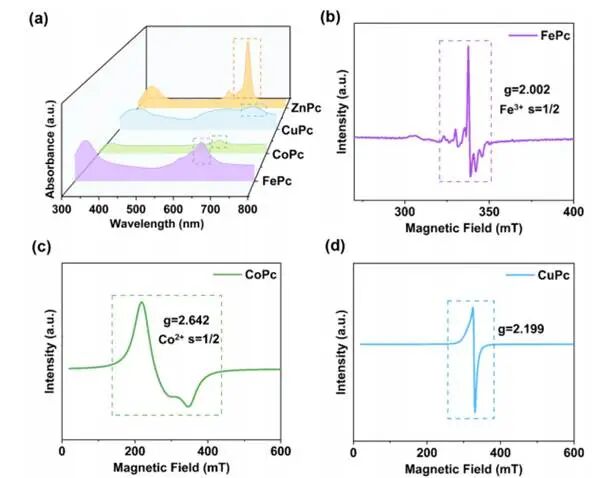
图3.a)FePc、CoPc、CuPc和ZnPc的紫外–可见吸收光谱;(b)FePc、(c)CoPc及(d)CuPc的电子顺磁共振(EPR)谱
通过记录UV-Vis吸光度谱和EPR谱图,证实了FePc、CoPc、CuPc和ZnPc电子结构中存在未配对电子。UV-Vis光谱显示600–750nm范围内存在Q带,该特征可归因于π–π*跃迁;四种过渡金属酞菁化合物中Q带峰的位置与强度差异则源于其d轨道上的未配对电子。Zn2+中没有未配对d电子的ZnPc呈现出强烈而尖锐的Q带,主要是由酞菁大环的π-π*跃迁引起的。相比之下,FePc、CoPc和CuPc的Q带因Fe3+、Co2+和Cu2+中的未配对d电子与酞菁分子的π环相互作用而减弱并展宽,这种相互作用导致自旋–轨道耦合作用,从而削弱了它们的Q带强度。
在EPR测试中,当材料中含有未配对电子时会出现特征峰。对于ZnPc而言,其Zn²⁺的电子结构中不存在未配对电子(因为所有d轨道均被完全占据),因此在ZnPc的EPR测量中无法检测到明显的顺磁信号或特征峰。而在FePc、CoPc和CuPc的EPR谱图中均观察到峰,表明中心金属离子上存在未配对电子。FePc样品的EPR谱在X波段显示典型信号,表明存在具有未配对电子的Fe³⁺;该信号对应的g值为2.002,可归属于酞菁分子M-N₄配位环境中的低自旋Fe³⁺中心,说明Fe³⁺的3dxz²轨道存在未配对电子。CoPc的EPR谱显示g值为2.642的信号,对应低自旋Co²⁺,表明Co²⁺的3dz²轨道存在未配对电子。CuPc的EPR谱则呈现Cu²⁺中心特有的轴向各向异性信号(g值为2.199),与CuPc中Cu²⁺(3d⁹)的典型EPR响应特征一致。
3.电化学性能
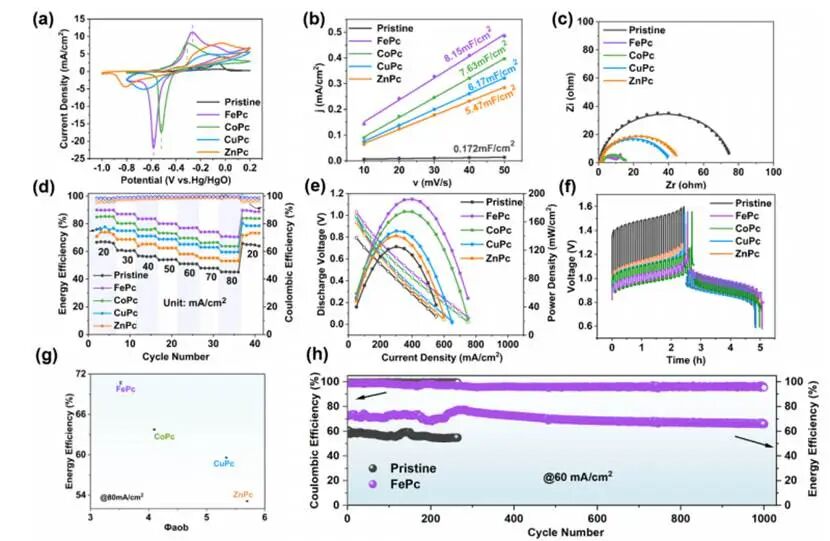
图4(a)原始玻碳电极及负载FePc、CoPc、CuPc和ZnPc的玻碳电极的循环伏安曲线;(b)原始电极、FePc、CoPc、CuPc及ZnPc电极的电流密度(j)与扫描速率(v)的线性拟合关系ECSA;(c)基于原始CF及负载FePc、CoPc、CuPc和ZnPc的CF电极电化学扫描谱(EIS)结果绘制的奈奎斯特图;(d)不同电流密度下原始CF及负载FePc、CoPc、CuPc和ZnPc的CF电极的EE与CE;(e)原始CF及负载FePc、CoPc、CuPc和ZnPc的CF电极的功率密度测试结果;(f)原始CF及负载FePc、CoPc、CuPc和ZnPc的CF电极的循环伏安(GITT)测试结果;(g)фaob值与平均EE的关系图;(h)原始CF与负载FePc的CF电极循环稳定性的对比分析
循环伏安法(CV)曲线显示原始玻碳未出现还原峰,表明S₄²⁻的还原反应难以在该材料上自发进行;而使用ZnPc和CuPc作为催化剂时,尽管存在还原峰,其电流密度均较低,分别为4.42mA/cm²(ZnPc)和5.13mA/cm²(CuPc);加入CoPc催化剂后,还原峰电流密度升至17.43mA/cm²;使用FePc时,还原峰电流密度进一步提升至21.96mA/cm²,表明FePc是这些催化剂中催化S₄²⁻还原反应最快的。ECSA曲线显示含ZnPc和CuPc的电极其Cdl分别仅为5.47mF/cm²和6.17mF/cm²;负载CoPc后,Cdl增至7.63mF/cm²;而负载FePc时Cdl达到最大值8.15mF/cm²,表明FePc具有最大的活性表面积。EIS测试显示FePc、CoPc、CuPc和ZnPc的溶液电阻(Rs)相近,分别为0.960Ω(FePc)、0.965Ω(CoPc)、0.961Ω(CuPc)和1.082Ω(ZnPc)。因此我们主要关注电荷转移电阻(Rct)的变化:无催化剂时Rct高达74.99Ω。当负载ZnPc和CuPc时,电荷转移过程加速,其Rct分别降至45.24Ω和39.14Ω。负载CoPc后,电荷转移过程进一步加速,Rct进一步降低至12.42Ω。当电极改性为FePc时,Rct值最小(9.86Ω),表明FePc加速了还原动力学过程。
全电池测试显示使用原始碳毡(CF)作为阳极,全电池在每个电流密度下的平均EE均小于70%,在80mA/cm2下,EE仅为45.18%。当负载ZnPc和CuPc时,EE略有提升,在80mA/cm²下分别为53.13%(ZnPc)和59.60%(CuPc);负载CoPc后,EE在80mA/cm²时升至63.74%。而负载FePc时EE显著提高,在20mA/cm²下达到89.61%,在80mA/cm²时仍保持70.70%。在功率密度测试中,与原始CF材料(125mW/cm²)相比,ZnPc和CuPc可使整电池的最大功率密度略有提升,分别达到140.34mW/cm²和147.17mW/cm²;负载CoPc时最大功率密度可达174.54mW/cm²;使用FePc时最大功率密度更可高达192.14mW/cm²。恒电流间歇滴定法测试表明:使用原始CF材料时,整电池在20 mA/cm²下的充电过电位高达512mV,放电过电位为123mV,证明从S4²⁻还原为S2²⁻的充电过程存在动力学迟缓现象;而在ZnPc、CuPc和CoPc催化作用下,充电过电位分别降至160mV、135mV和107mV;FePc的催化作用更将充电过电位大幅降低至51.1mV,加速了S4²⁻向S2²⁻的还原反应动力学。

本研究成功揭示了过渡金属基酞菁中催化中心的电子结构在加速电化学还原S42-的缓慢动力学中的性质。研究结果表明在催化中心与目标反应物之间的轨道对称性匹配且存在未配对d电子的条件下,催化活性明显更为优异。基于上述研究成果,作者团队构建了一个包含催化过程四个关键步骤的定量描述模型,该模型直接揭示了催化剂电子结构与其催化活性之间的关联性。理论预测显示具有最优电子结构的FePc催化剂在80mA/cm²下可实现高达70.70%的EE,展现出卓越的催化性能。